The human voltage gated potassium channel Kv1.5 that conducts the IKur current is a key determinant of the atrial action potential. ThIts mutatis channel is an attractive target for the management of Aons have been linked to hereditary forms of atrial Ffibrillation (AF). A wide range of peptide toxins from venomous animals are targeting ion , and the channels, including mammalian channels. These peptides usually have a much larger interacting surface with the ion channel compared to small molecule inhibitors and thus, generally confer higher selectivity to the peptide is an attractive target for the management of AF. The development of IKur blockers. To date, literature has known two peptides that to treat AF resulted in small molecule Kv1.5 inhibit IKur: Ts6 and Osu1. Tors.
- Kv1.5
- IKur
- peptide inhibitor
- atrial fibrillation
1. Diseases Related to Kv1.5
2. Atrial Fibrillation and Possible Pharmacological Treatments
- -
-
Development and improvement of existing antiarrhythmic agents: Amiodarone derivates, Multi-channel blockers, etc.
- -
-
Atrial selective therapeutic agents (ARDA): IKur blocker; IK,Ach blocker; INa, IKr blockers
- -
-
Upstream therapy agents, drugs affecting: structural remodeling,; inflammation,; hypertrophy,; oxidative stress,; etc.,
- -
-
Gap junction modulators: Antiarrhythmic peptides affecting connexins Cx40 and Cx43
3. Peptide Modulators of the Kv Channels

3.1. Pore Blocker Peptides
Mechanism of Action of the Pore Blockers
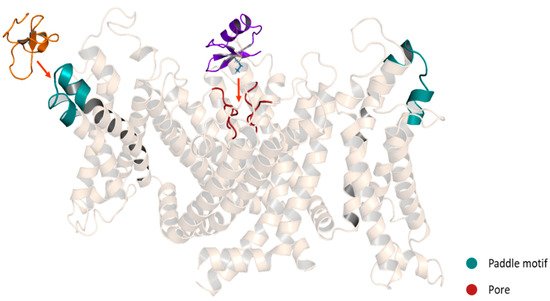
3.2. Gating Modifiers Peptides
Mechanism of Action of the Gating Modifiers
4. Osu1 and Ts6: The Known Peptide Modulators of the Kv1.5
References
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191.
- Christophersen, I.E.; Olesen, M.S.; Liang, B.; Andersen, M.N.; Larsen, A.P.; Nielsen, J.B.; Haunsø, S.; Olesen, S.-P.; Tveit, A.; Svendsen, J.H.; et al. Genetic variation in KCNA5: Impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Heart J. 2013, 34, 1517–1525.
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations ofKCNA5in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol.-Cell Physiol. 2007, 292, C1837–C1853.
- Fu, L.; Lv, Y.; Zhong, Y.; He, Q.; Liu, X.; Du, L. Tyrosine phosphorylation of Kv1.5 is upregulated in intrauterine growth retardation rats with exaggerated pulmonary hypertension. Braz. J. Med. Biol. Res. 2017, 50, e6237.
- Macfarlane, S.N.; Sontheimer, H. Modulation of Kv1.5 Currents by Src Tyrosine Phosphorylation: Potential Role in the Differentiation of Astrocytes. J. Neurosci. 2000, 20, 5245–5253.
- Preussat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wölfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1.3 and Kv1.5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36.
- Bielanska, J.; Hernandez-Losa, J.; Perez-Verdaguer, M.; Moline, T.; Somoza, R.; Cajal, S.R.Y.; Condom, E.; Ferreres, J.C.; Felipe, A. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets 2009, 9, 904–914.
- Vallejo-Gracia, A.; Bielanska, J.; Hernández-Losa, J.; Castellví, J.; Ruiz-Marcellan, M.C.; Cajal, S.R.Y.; Condom, E.; Manils, J.; Soler, C.; Comes, N.; et al. Emerging role for the voltage-dependent K+ channel Kv1.5 in B-lymphocyte physiology: Expression associated with human lymphoma malignancy. J. Leukoc. Biol. 2013, 94, 779–789.
- Chung, M.K.; Eckhardt, L.L.; Chen, L.Y.; Ahmed, H.M.; Gopinathannair, R.; Joglar, J.A.; Noseworthy, P.A.; Pack, Q.R.; Sanders, P.; Trulock, K.M. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement from the American Heart Association. Circulation 2020, 141, e750–e772.
- Conen, D. Epidemiology of atrial fibrillation. Eur. Heart J. 2018, 39, 1323–1324.
- Guo, X.; Chen, W.; Sun, H.; You, Q. Kv1.5 Inhibitors for Treatment of Atrial Fibrillation: A Tradeoff between Selectivity and Non-selectivity. Curr. Top. Med. Chem. 2016, 16, 1843–1854.
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and Molecular Relationships of Voltage-Gated Potassium Channels. Pharmacol. Rev. 2005, 57, 473–508.
- Wettwer, E.; Terlau, H. Pharmacology of voltage-gated potassium channel Kv1.5—Impact on cardiac excitability. Curr. Opin. Pharmacol. 2014, 15, 115–121.
- Attali, B.; Chandy, K.G.; Giese, M.H.; Grissmer, S.; Gutman, G.A.; Jan, L.Y.; Lazdunski, M.; McKinnon, D.; Nerbonne, J.; Pardo, L.A.; et al. Voltage-gated potassium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019, 2019.
- Bajaj, S.; Han, J. Venom-Derived Peptide Modulators of Cation-Selective Channels: Friend, Foe or Frenemy. Front. Pharmacol. 2019, 10, 58.
- Wirth, K.J.; Paehler, T.; Rosenstein, B.; Knobloch, K.; Maier, T.; Frenzel, J.; Brendel, J.; Busch, A.E.; Bleich, M. Atrial effects of the novel K+-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc. Res. 2003, 60, 298–306.
- Christ, T.; Wettwer, E.; Voigt, N.; Hála, O.; Radicke, S.; Matschke, K.; Várro, A.; Dobrev, D.; Ravens, U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br. J. Pharmacol. 2008, 154, 1619–1630.
- Wettwer, E.; Hála, O.; Christ, T.; Heubach, J.F.; Dobrev, D.; Knaut, M.; Varró, A.; Ravens, U. Role of IKur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 2004, 110, 2299–2306.
- Wirth, K.J.; Steinmeyer, K.; Ruetten, H. Sensitization of Upper Airway Mechanoreceptors as a New Pharmacologic Principle to Treat Obstructive Sleep Apnea: Investigations with AVE0118 in Anesthetized Pigs. Sleep 2013, 36, 699–708.
- Shunmugam, S.R.; Sugihara, C.; Freemantle, N.; Round, P.; Furniss, S.; Sulke, N. A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers allowing continuous beat-to-beat monitoring of drug efficacy. J. Interv. Card. Electrophysiol. 2018, 51, 191–197.
- Lagrutta, A.; Wang, J.; Fermini, B.; Salata, J.J. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063.
- Fedida, D.; Orth, P.M.; Chen, J.Y.; Lin, S.; Plouvier, B.; Jung, G.; Ezrin, A.M.; Beatch, G.N. The Mechanism of Atrial Antiarrhythmic Action of RSD1235. J. Cardiovasc. Electrophysiol. 2005, 16, 1227–1238.
- Wettwer, E.; Christ, T.; Endig, S.; Rozmaritsa, N.; Matschke, K.; Lynch, J.J.; Pourrier, M.; Gibson, J.K.; Fedida, D.; Knaut, M.; et al. The new antiarrhythmic drug vernakalant: Ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc. Res. 2013, 98, 145–154.
- Dobrev, D.; Hamad, B.; Kirkpatrick, P. Vernakalant. Nat. Rev. Drug Discov. 2010, 9, 915–916.
- Burashnikov, A.; Pourrier, M.; Gibson, J.K.; Lynch, J.J.; Antzelevitch, C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel: Comparison with ranolazine and dl-sotalol. Circ. Arrhythmia Electrophysiol. 2012, 5, 400–408.
- van Middendorp, L.B.; Strik, M.; Houthuizen, P.; Kuiper, M.; Maessen, J.G.; Auricchio, A.; Prinzen, F.W. Electrophysiological and haemodynamic effects of vernakalant and flecainide in dyssynchronous canine hearts. Europace 2014, 16, 1249–1256.
- Camm, A.J.; Capucci, A.; Hohnloser, S.H.; Torp-Pedersen, C.; Van Gelder, I.C.; Mangal, B.; Beatch, G. A Randomized Active-Controlled Study Comparing the Efficacy and Safety of Vernakalant to Amiodarone in Recent-Onset Atrial Fibrillation. J. Am. Coll. Cardiol. 2011, 57, 313–321.
- Cialdella, P.; Pedicino, D.; Santangeli, P. Novel Agents for the Acute Conversion of Atrial Fibrillation: Focus on Vernakalant. Recent Patents Cardiovasc. Drug Discov. 2011, 6, 1–8.
- Simon, A.; Niederdoeckl, J.; Janata, K.; Spiel, A.O.; Schuetz, N.; Schnaubelt, S.; Herkner, H.; Cacioppo, F.; Laggner, A.N.; Domanovits, H. Vernakalant and electrical cardioversion for AF–Safe and effective. IJC Heart Vasc. 2019, 24, 100398.
- Hall, A.J.; Mitchell, A. Introducing Vernakalant into Clinical Practice. Arrhythmia Electrophysiol. Rev. 2019, 8, 70–74.
- Bergeron, Z.L.; Bingham, J.P. Scorpion toxins specific for potassium (K+) channels: A historical overview of peptide bioengineering. Toxins 2012, 4, 1082–1119.
- Rash, L.; Hodgson, W.C. Pharmacology and biochemistry of spider venoms. Toxicon 2002, 40, 225–254.
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276.
- Jungo, F.; Bairoch, A. Tox-Prot, the toxin protein annotation program of the Swiss-Prot protein knowledgebase. Toxicon 2005, 45, 293–301.
- Jungo, F.; Estreicher, A.; Bairoch, A.; Bougueleret, L.; Xenarios, I. Animal Toxins: How is Complexity Represented in Databases? Toxins 2010, 2, 262–282.
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557.
- Quintero-Hernández, V.; Jiménez-Vargas, J.; Gurrola, G.; Valdivia, H.; Possani, L. Scorpion venom components that affect ion-channels function. Toxicon 2013, 76, 328–342.
- Kuzmenkov, A.I.; Krylov, N.; Chugunov, A.O.; Grishin, E.V.; Vassilevski, A.A. Kalium: A database of potassium channel toxins from scorpion venom. Database 2016, 2016, baw056.
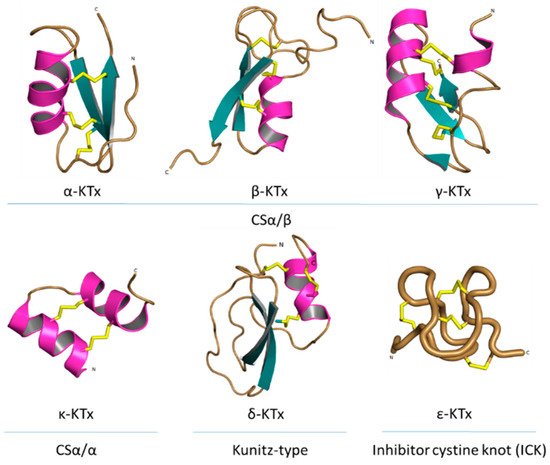
- Tytgat, J.; Chandy, K.G.; Garcia, M.L.; Gutman, G.A.; Martin-Eauclaire, M.F.; van der Walt, J.J.; Possani, L.D. A unified nomenclature for short-chain peptides isolated from scorpion venoms: Alpha-KTx molecular subfamilies. Trends Pharmacol. Sci. 1999, 20, 444–447.
- de la Vega, R.C.R.; Possani, L.D. Current views on scorpion toxins specific for K+-channels. Toxicon 2004, 43, 865–875.
- Zhu, S.; Gao, B.; Aumelas, A.; Rodríguez, M.D.C.; Lanz-Mendoza, H.; Peigneur, S.; Diego-Garcia, E.; Martin-Eauclaire, M.-F.; Tytgat, J.; Possani, L.D. MeuTXKβ1, a scorpion venom-derived two-domain potassium channel toxin-like peptide with cytolytic activity. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 872–883.
- Kuzmenkov, A.I.; Grishin, E.V.; Vassilevski, A.A. Diversity of Potassium Channel Ligands: Focus on Scorpion Toxins. Biochemistry 2015, 80, 1764–1799.
- Bartok, A.; Panyi, G.; Varga, Z. Potassium Channel Blocking Peptide Toxins from Scorpion Venom. In Scorpion Venoms; Springer: Dordrecht, The Netherlands, 2015; pp. 493–527.
- Diego-García, E.; Schwartz, E.F.; D’Suze, G.; González, S.A.R.; Batista, C.V.; García, B.I.; de la Vega, R.C.R.; Possani, L.D. Wide phylogenetic distribution of Scorpine and long-chain beta-KTx-like peptides in scorpion venoms: Identification of “orphan” components. Peptides 2007, 28, 31–37.
- Jiménez-Vargas, J.; Restano-Cassulini, R.; Possani, L. Toxin modulators and blockers of hERG K+ channels. Toxicon 2012, 60, 492–501.
- Corona, M.; Gurrola, G.B.; Merino, E.; Cassulini, R.R.; Valdez-Cruz, N.A.; García, B.; Ramírez-Domínguez, M.E.; Coronas, F.I.; Zamudio, F.Z.; Wanke, E.; et al. A large number of novel Ergtoxin-like genes and ERG K+-channels blocking peptides from scorpions of the genus Centruroides. FEBS Lett. 2002, 532, 121–126.
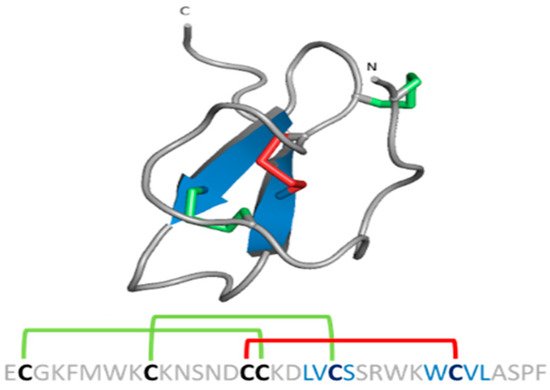
- Chagot, B.; Pimentel, C.; Dai, L.; Pil, J.; Tytgat, J.; Nakajima, T.; Corzo, G.; Darbon, H.; Ferrat, G. An unusual fold for potassium channel blockers: NMR structure of three toxins from the scorpion Opisthacanthus madagascariensis. Biochem. J. 2005, 388, 263–271.
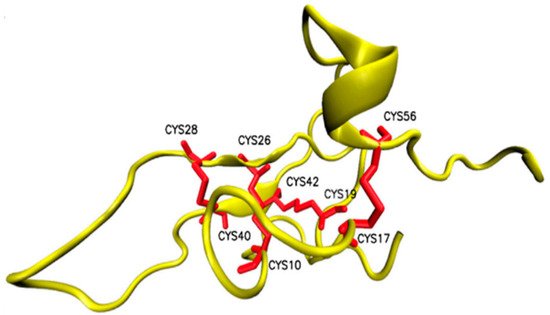
- Zhao, R.; Dai, H.; Qiu, S.; Li, T.; He, Y.; Ma, Y.; Chen, Z.; Wu, Y.; Li, W.; Cao, Z. SdPI, The First Functionally Characterized Kunitz-Type Trypsin Inhibitor from Scorpion Venom. PLoS ONE 2011, 6, e27548.
- Chen, Z.-Y.; Hu, Y.-T.; Yang, W.-S.; He, Y.-W.; Feng, J.; Wang, B.; Zhao, R.-M.; Ding, J.-P.; Cao, Z.-J.; Li, W.-X.; et al. Hg1, Novel Peptide Inhibitor Specific for Kv1.3 Channels from First Scorpion Kunitz-type Potassium Channel Toxin Family. J. Biol. Chem. 2012, 287, 13813–13821.
- Cremonez, C.M.; Maiti, M.; Peigneur, S.; Cassoli, J.S.; Dutra, A.A.A.; Waelkens, E.; Lescrinier, E.; Herdewijn, P.; De Lima, M.E.; Pimenta, A.M.C.; et al. Structural and Functional Elucidation of Peptide Ts11 Shows Evidence of a Novel Subfamily of Scorpion Venom Toxins. Toxins 2016, 8, 288.
- Jiménez-Vargas, J.M.; Possani, L.D.; Luna-Ramírez, K. Arthropod toxins acting on neuronal potassium channels. Neuropharmacology 2017, 127, 139–160.
- Gao, B.; Harvey, P.J.; Craik, D.J.; Ronjat, M.; De Waard, M.; Zhu, S. Functional evolution of scorpion venom peptides with an inhibitor cystine knot fold. Biosci. Rep. 2013, 33, 513–527.
- Banerjee, A.; Lee, A.; Campbell, E.B.; MacKinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2013, 2, e00594.
- Mouhat, S.; Mosbah, A.; Visan, V.; Wulff, H.; Delepierre, M.; Darbon, H.; Grissmer, S.; De Waard, M.; Sabatier, J.-M. The functional dyad of scorpion toxin Pi1 is not itself a prerequisite for toxin binding to the voltage-gated Kv1.2 potassium channels. Biochem. J. 2004, 377, 25–36.
- Cui, M.; Shen, J.; Briggs, J.M.; Fu, W.; Wu, J.; Zhang, Y.; Luo, X.; Chi, Z.; Ji, R.; Jiang, H.; et al. Brownian dynamics simulations of the recognition of the scorpion toxin P05 with the small-conductance calcium-activated potassium channels. J. Mol. Biol. 2002, 318, 417–428.
- Pardo-Lopez, L.; Zhang, M.; Liu, J.; Jiang, M.; Possani, L.D.; Tseng, G.N. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ErgTx) to the channel’s outer vestibule. J. Mol. Biol. 2002, 277, 16403–16411.
- Li-Smerin, Y.; Swartz, K.J. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl. Acad. Sci. USA 1998, 95, 8585–8589.
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498.
- Redaelli, E.; Cassulini, R.R.; Silva, D.F.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagón, A.; et al. Target Promiscuity and Heterogeneous Effects of Tarantula Venom Peptides Affecting Na+ and K+ Ion Channels. J. Biol. Chem. 2010, 285, 4130–4142.
- Diochot, S.; Drici, M.-D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263.
- Park, J.H.; Carlin, K.P.; Wu, G.; Ilyin, V.I.; Musza, L.L.; Blake, P.R.; Kyle, D.J. Studies Examining the Relationship between the Chemical Structure of Protoxin II and Its Activity on Voltage Gated Sodium Channels. J. Med. Chem. 2014, 57, 6623–6631.
- Escoubas, P.; Diochot, S.; Célérier, M.L.; Nakajima, T.; Lazdunski, M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol. Pharmacol. 2002, 62, 48–57.
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 2016, 534, 494–499.
- Montandon, G.G.; Cassoli, J.S.; Peigneur, S.; Verano-Braga, T.; dos Santos, D.M.; Paiva, A.L.B.; de Moraes, É.R.; Kushmerick, C.; Borges, M.H.; Richardson, M.; et al. GiTx1(β/κ-theraphotoxin-Gi1a), a novel toxin from the venom of Brazilian tarantula Grammostola iheringi (Mygalomorphae, Theraphosidae): Isolation, structural assessments and activity on voltage-gated ion channels. Biochimie 2020, 176, 138–149.
- Lee, S.-Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235.
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S. SGTx1, a Kv Channel Gating-Modifier Toxin, Binds to the Interfacial Region of Lipid Bilayers. Biophys. J. 2007, 92, L07–L09.
- Wang, Y.; Luo, Z.; Lei, S.; Li, S.; Li, X.; Yuan, C. Effects and mechanism of gating modifier spider toxins on the hERG channel. Toxicon 2021, 189, 56–64.
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-Venom Peptides as Therapeutics. Toxins 2010, 2, 2851–2871.
- Colgrave, M.L.; Craik, D.J. Thermal, Chemical, and Enzymatic Stability of the Cyclotide Kalata B1: The Importance of the Cyclic Cystine Knot. Biochemistry 2004, 43, 5965–5975.
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230.
- Ozawa, S.-I.; Kimura, T.; Nozaki, T.; Harada, H.; Shimada, I.; Osawa, M. Structural basis for the inhibition of voltage-dependent K+ channel by gating modifier toxin. Sci. Rep. 2015, 5, 14226.
- Li-Smerin, Y.; Swartz, K.J. Localization and Molecular Determinants of the Hanatoxin Receptors on the Voltage-Sensing Domains of a K+ Channel. J. Gen. Physiol. 2000, 115, 673–684.
- Tao, H.; Chen, X.; Deng, M.; Xiao, Y.; Wu, Y.; Liu, Z.; Zhou, S.; He, Y.; Liang, S. Interaction site for the inhibition of tarantula Jingzhaotoxin-XI on voltage-gated potassium channel Kv2.1. Toxicon 2016, 124, 8–14.
- DeSimone, C.V.; Lu, Y.; Bondarenko, V.E.; Morales, M.J. S3b Amino Acid Substitutions and Ancillary Subunits Alter the Affinity of Heteropoda venatoria Toxin 2 for Kv4.3. Mol. Pharmacol. 2009, 76, 125–133.
- Ebbinghaus, J.; Legros, C.; Nolting, A.; Guette, C.; Celerier, M.-L.; Pongs, O.; Bähring, R. Modulation of Kv4.2 channels by a peptide isolated from the venom of the giant bird-eating tarantula Theraphosa leblondi. Toxicon 2004, 43, 923–932.
- Alvarado, D.; Cardoso-Arenas, S.; Corrales-García, L.-L.; Clement, H.; Arenas, I.; Montero-Dominguez, P.A.; Olamendi-Portugal, T.; Zamudio, F.; Csoti, A.; Borrego, J.; et al. A Novel Insecticidal Spider Peptide that Affects the Mammalian Voltage-Gated Ion Channel hKv1.5. Front. Pharmacol. 2021, 11, 1937.
- Cerni, F.A.; Pucca, M.B.; Peigneur, S.; Cremonez, C.M.; Bordon, K.C.F.; Tytgat, J.; Arantes, E.C. Electrophysiological Characterization of Ts6 and Ts7, K+ Channel Toxins Isolated through an Improved Tityus serrulatus Venom Purification Procedure. Toxins 2014, 6, 892–913.
- Martin-Eauclaire, M.F.; Pimenta, A.M.; Bougis, P.E.; De Lima, M.E. Potassium channel blockers from the venom of the Brazilian scorpion Tityus serrulatus (Lutz and Mello, 1922). Toxicon 2016, 119, 253–265.
- Zoccal, K.F.; da Silva Bitencourt, C.; Sorgi, C.A.; Bordon, K.D.C.F.; Sampaio, S.V.; Arantes, E.C.; Faccioli, L.H. Ts6 and Ts2 from Tityus serrulatus venom induce inflammation by mechanisms dependent on lipid mediators and cytokine production. Toxicon 2013, 61, 1–10.