Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Anke Van den Berg.
Bacillus thuringiensis (Bt) is a bacterium capable of producing Cry toxins, which are recognized for their bio-controlling actions against insects. However, a few Bt strains encode proteins lacking insecticidal activity but showing cytotoxic activity against different cancer cell lines and low or no cytotoxicity toward normal human cells.
- parasporins
- Bacillus thuringiensis
- anti-cancer
1. Background
Bacillus thuringiensis (Bt) is an endospore-forming aerobic bacterium with a high capacity to resist elevated temperatures and desiccation conditions, characterized by producing parasporal toxins [1]. Bt was first identified in 1901 by Shigetane Ishiwata, who reported that this microorganism had an infective capacity toward Bombyx mori. This plague caused severe damage to the silk industry in Japan [2]. At that time, the author termed the bacteria Bacillus sotto. A decade later, Berliner isolated a Gram-positive bacterium in Ephesitia kuehniella larvae in the state of Thuringia (Germany). Ignoring the identification given by Ishiwata, Berliner designated the bacterium as Bacillus thuringiensis, and this nomenclature has remained ever since [3].
Bt toxins, such as the Cry and Cytolytic (Cyt) proteins, are present in crystal form, with toxic effects in several pest vectors. A subset of the Cry proteins present in the crystals are parasporins (PSs), proteins with cytotoxic activity in human cancer cell lines [4]. The wide spectrum of the potential applications of PSs in biotechnological and biological medicine research has made Bt one of the most essential microorganisms used as a bio-controller and, more recently, as a producer of non-insecticidal parasporal proteins [5].
2. Overview of the Classification and Structure of Parasporins Found in Bacillus thuringiensis Strains
In the search for new therapeutic agents to treat cancer, bacterial proteins have become a focus of attention in the last two decades. In 1999, Mizuki et al. conducted a bioprospecting study using around 1700 Bt isolates by selecting 42 candidates with no hemolytic and cytotoxic activity to test their activity against MOLT-4 cells (human lymphoblastic leukemia) [6,7][6][7]. Prasad, Seki, and their teams described the existence of a 13-kDa parasporal toxin from a Bt strain with dual activity, i.e., insecticidal action against Bombyx mori and antitumor activity in colon and blood cancer cell lines [1,8][1][8]. This new promising protein, termed a parasporin, is among the Bt proteins with biological activities that potentially allow for medical applications.
The Parasporin Classification and Nomenclature Committee defined the term “parasporins” in 2006 as “parasporal proteins of Bt and related bacteria that are non-hemolytic but are preferentially able to kill cancer cells” [9]. To date, six parasporin families (PS1–PS6) including 19 PSs produced by at least 11 Bt strains have been identified mainly in four countries (Japan, Vietnam, India, and Canada) (Table 1) [10,11][10][11]. PSs are divided into those of higher molecular mass (PS1, PS3, and PS6), approximately 80 kDa, which are processed into active 60 kDa molecules, and those with lower molecular mass (PS2, PS4, and PS5), originated from precursors of 33 to 37 kDa, which are processed by proteolytic cleavage to 30 kDa molecules. When a proteolytic cleavage is made by a serine protease, such as Proteinase K, at the C- terminal and N- terminal residues of the precursor, the toxin of 60 kDa and 30 kDa is active with cytocidal activity [9,10,12][9][10][12]. All the PS family members are characterized by a conserved structure consisting of three domains (Figure 1A, B). A more detailed structural and sequence analysis is presented by Xu et al. [9].
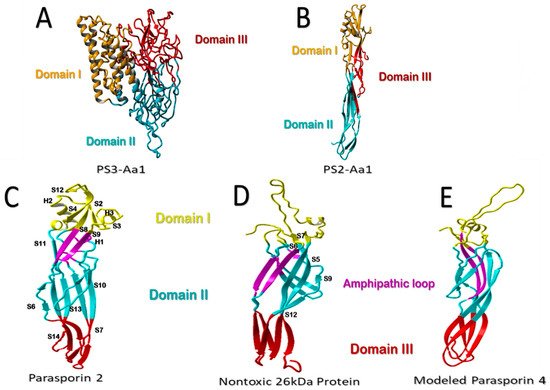
Figure 1. Structural comparison of parasporins. (A) Structural model of higher-molecular-weight PS3Aa1 with its three domains. (B) Low-molecular-weight PS2Aa1 structural model. (C–E) Structural comparison between parasporin-2, the 26-kDa nontoxic protein, and aerolysin-like β-PFT. Membrane-binding-related domain I is colored yellow. The membrane-insertion and pore-formation regions are colored blue (domain II) and red (Domain III). It is suggested that the purple amphipathic β-hairpin is necessary for pore formation (C–E). Parasporin 4 (PS4) was modeled using the 26-kDa nontoxic protein as an adapted template from Xu et al. [9], modified by the authors.
3. Effects of Parasporins on Cancer Cells
The mechanism of action of pore-forming proteins (PFPs) is dynamic, with three main steps: (1) the formation of water solubility, (2) self-assembly, and (3) insertion into the membrane, which leads to a pore suspected to be highly destructive for membrane integrity [34][13]. The points at which these proteins anchor to the membrane probably occur at specific receptors located in the microdomains rich in cholesterol and sphingolipids (lipid rafts), since these are requirements for GPI-anchored proteins, and the glucan region may be required for the binding and assembly in the membrane (Figure 2, part 1) [9]. Similarly, it was reported that the cell membrane receptor Beclin-1 could be important in the binding of three-domain parasporins (Parts 2 and 3, Figure 2) and that the Beclin-1 receptor is present in the mammary epithelium and epithelial carcinoma cells (Figure 2) [9,34,35][9][13][14].
The rearrangement of the domains typical for the classic protein model of three pore-forming domains does not occur for PS1 [36][15]. Therefore, its activity is not oriented to forming pores in the membrane [21,36][16][15]. PS1 was proposed to function as an activator of the apoptotic signaling pathway [14,19,37][17][18][19]. Selective cytotoxicity has been reported for the HeLa, Sawano, HepG2, HL-60, and MOLT-4 cell lines after PS1’s proteolytic activation by trypsin (Table 1) [6,38][6][20]. The activity of PS1 mainly involves modulating the influx of Ca2+ levels [6,21,39][6][16][21].
The PS2 mechanism of action likely starts with recognizing and binding to a receptor located in the cancer cells’ membranes [24][22], identifying lipid rafts, and anchoring the protein monomers in the periphery. The oligomers, resistant to sodium dodecyl sulfate (SDS), are embedded in the membrane, leading to its permeabilization [18,24][23][22]. Although PS2 is considered a selective pore-forming toxin, its primary receptors have not been fully elucidated [18][23].
Cells exposed to PS2 show morphological changes, including inflammation, blisters and lysis, microtubule disassembly, actin filament coiling, and fragmentation of the mitochondria and endoplasmic reticulum. PS2 resides in the plasma membrane and has been shown to activate apoptosis through caspases [14][17], triggering increased permeability [14,18,31][17][23][24]. These effects are induced by the accumulation of PS2 by large oligomers in the membrane’s lipid rafts [8,18,24,39][8][23][22][21]. In turn, the association of PS2 with GPI is required for cytolytic action. By contrast, membrane cholesterol slightly affects the efficiency of oligomerization [1]. The activation of PS2 induced by proteinase K [25] leads to the exposure of specific regions that bind to the receptor [18,25][23][25].
PS3 acts as a pore former in cancer cells, thereby increasing cellular permeability [12,40][12][26]. Although PS3 is structurally similar to the Cry proteins, containing the five conserved blocks that characterize Cry [7[7][26],40], the PS3 and Cry proteins are fundamentally different due to a castor domain [7,40[7][26][27],41], which is present in many unrelated proteins and is presumed to enhance/induce carbohydrate-binding capacity [40][26]. Similar to the above-described PS, the mechanisms of action of PS3 remain largely unknown. Krishnan et al. suggested that PS3 is most likely pore forming [16][28], which leads to an imbalance in ATP, increased cell size, and membrane damage [40,41][26][27]. Its cytotoxic activity was evident in the HL-60 and HepG2 cell lines [7[7][26][27],40,41], but it did not affect HeLa cells [41][27].
Studies on PS3, PS4, PS5, and PS6 are limited compared to those on PS1 and PS2, and many action modes remain undetermined. PS4 shows homology with both Cry and pore-forming β-type aerolysin. It has been reported to be cytotoxic to the Caco-2, Sawano, and MOLT cell lines [5,41][5][27]. Its structure mainly comprises β-sheet domains, and its pore-forming activity is not dependent on cholesterol [21,41][16][27]. Cells treated with this protein show an increase in size due to an increase in the cytoplasmic compartment and shrinkage of the nucleus, leading to the rupture of the cytoplasmic membrane and cell death [42][29]. PS5 and PS6 are the most recently discovered PS proteins. They have three domains, similar to PS1 and PS2, and presumably have pore-forming activity. They have been reported to show cytotoxic activity in liver and cervical cancer cell lines. However, there is no further information on their mechanisms of action [14][17].
4. Perspectives on the Improvement of Bt Parasporins as an Innovative Strategy for Controlling Cancer Cells
By deciphering structure-function relationships, proteins with improved properties, e.g., desired thermal activity, selectivity, specificity, or folding, can be designed [43][30]. For example, engineered proteins with various substitutions of amino acids are used in receptor- and channel-protein-binding studies [44][31]. Protein engineering is called the synthesis of proteins with enhanced functionality in vitro and in vivo due to altered physical, chemical, or biological properties through genomic and post-genomic strategies. Genetic improvement is closely linked to complementary computational methods, which aim to optimize the generation of mutant libraries by simulating the experimental conditions of directed mutagenesis techniques [45,46,47,48][32][33][34][35]. In addition, other computational methods are oriented toward predicting protein structures and designing models that allow the prediction of molecular interactions and pinpoint amino-acid residues or regions at crucial positions in natural and mutant proteins [43,49][30][36]. The computational technique most widely used for studying the possible interactions of Bt Cry toxins with insect receptors is molecular docking, followed by molecular dynamics, which has proven to help predict the stability of the interactions and analyze the molecular mechanisms of action. Florez et al. [50][37] obtained five Cry11 variants by DNA shuffling and showed the toxic activity against Aedes aegypti and Culex quinquefasciatus for three of them. Molecular docking simulations were performed for these three variants, and the amino acids with possible interactions were identified. BenFarhat-Touzri et al. [51][38] cloned and sequenced the Cry1D-133 toxin and determined its toxicity against S. littoralis larvae. Molecular docking simulations were performed to explain the enhanced toxicity of this toxin and showed that the number of toxin–receptor interactions was higher than that of the interactions exhibited by the Cry1D toxin. The use of computational techniques based on molecular dynamics has enabled researchers to study the mechanisms of action of Cry toxins. The study of molecular dynamics has provided novel insights into the oligomerization of Cry toxins at a molecular level. Sriwimol et al. simulated the Cry4Ba structure with a three-dimensional reconstructed map for trimeric protein states. For the first time, they showed the need for membrane-induced conformational changes in Cry4Ba toxin monomers to allow the molecular assembly of a pre-pore trimer, which can be inserted into the target membranes to generate a lytic pore [52][39]. Other molecular dynamic studies have been applied to investigate the residue interactions relevant to the toxicity of the Bt Cry toxin family. Pacheco et al. discovered the importance of salt-bridge formation between α-helix residues from adjacent monomers for the toxicity and oligomerization of the Cry1Ab and Cry5Ba toxins by molecular dynamics’ simulations [53][40]. They showed a critical role for the salt bridge between the E101 and R99 residues of Cry1Ab [54][41]. Site-directed mutagenesis experiments confirmed decreased oligomerization and toxicity potential for Cry1Ab-E101K and Cry1Ab-R99E mutants. Interestingly, the R99–E101 salt bridge is not fully conserved in Cry proteins, with both or one of the residues being different in Cry5Ba. However, Pacheco et al. showed that additional salt bridges with similar structural functions could also be formed in these Cry proteins. In conclusion, the computational analysis highlighted the importance of salt-bridge formation between the α-3 helices of adjacent monomers for inducing/facilitating a conformational change crucial for Cry toxicity [53][40].References
- Portela-Dussán, D.D.; Chaparro-Giraldo, A.; López-Pazos, S.A. Bacillus thuringiensis biotechnology in agriculture. Nova 2013, 11, 87–96.
- Melo, A.L.D.A.; Soccol, V.T.; Soccol, C.R. Bacillus thuringiensis: Mechanism of action, resistance, and new applications: A review. Crit. Rev. Biotechnol. 2016, 36, 317–326.
- Akao, T.; Mizuki, E.; Yamashita, S.; Kim, H.S.; Lee, D.W.; Ohba, M. Specificity of lectin activity of Bacillus thuringiensis parasporal inclusion proteins. J. Basic Microbiol. 2001, 41, 3–6.
- Akiba, T.; Okumura, S. Parasporins 1 and 2: Their structure and activity. J. Invertebr. Pathol. 2017, 142, 44–49.
- Mizuki, E.; Ohba, M.; Akao, T.; Yamashita, S.; Saitoh, H.; Park, Y.S. Unique activity associated with non-insecticidal Bacillus thuringiensis parasporal inclusions: In vitro cell-killing action on human cancer cells. J. Appl. Microbiol. 1999, 86, 477–486.
- Ammons, D.R.; Short, J.D.; Bailey, J.; Hinojosa, G.; Tavarez, L.; Salazar, M.; Rampersad, J.N. Anti-cancer Parasporin toxins are associated with different environments: Discovery of two novel Parasporin 5-like genes. Curr. Microbiol. 2016, 72, 184–189.
- Moazamian, E.; Bahador, N.; Azarpira, N.; Rasouli, M. Anti-cancer Parasporin toxins of new Bacillus thuringiensis against human colon (HCT-116) and blood (CCRF-CEM) cancer cell lines. Curr. Microbiol. 2018, 75, 1090–1098.
- Xu, C.; Wang, B.C.; Yu, Z.; Sun, M. Structural insights into Bacillus thuringiensis Cry, Cyt and parasporin toxins. Toxins 2014, 6, 2732–2770.
- Kitada, S.; Abe, Y.; Shimada, H.; Kusaka, Y.; Matsuo, Y.; Katayama, H.; Okumura, S.; Akao, T.; Mizuki, E.; Kuge, O.; et al. Cytocidal actions of parasporin-2, an anti-tumor crystal toxin from Bacillus thuringiensis. J. Biol. Chem. 2006, 281, 26350–26360.
- Gonzalez, E.; Granados, J.C.; Short, J.D.; Ammons, D.R.; Rampersad, J. Parasporins from a Caribbean Island: Evidence for a globally dispersed Bacillus thuringiensis strain. Curr. Microbiol. 2011, 62, 1643–1648.
- Brasseur, K.; Auger, P.; Asselin, E.; Parent, S.; Cote, J.C.; Sirois, M. Parasporin-2 from a new Bacillus thuringiensis 4R2 strain induces caspases activation and apoptosis in human cancer cells. PLoS ONE 2015, 10, e0135106.
- Mizuki, E.; Park, Y.S.; Saitoh, H.; Yamashita, S.; Akao, T.; Higuchi, K.; Ohba, M. Parasporin, a human leukemic cell-recognizing parasporal protein of Bacillus thuringiensis. Clin. Diagn. Lab. Immunol. 2000, 7, 625–634.
- Prasad, S.S.; Shethna, Y.I. Purification, crystallization and partial characterization of the antitumour and insecticidal protein subunit from the delta-endotoxin of Bacillus thuringiensis var. thuringiensis. Biochim. Biophys. Acta 1974, 362, 558–566.
- Patyar, S.; Joshi, R.; Byrav, D.P.; Prakash, A.; Medhi, B.; Das, B. Bacteria in cancer therapy: A novel experimental strategy. J. Biomed. Sci. 2010, 17, 21.
- Katayama, H.; Yokota, H.; Akao, T.; Nakamura, O.; Ohba, M.; Mekada, E.; Mizuki, E. Parasporin-1, a novel cytotoxic protein to human cells from non-insecticidal parasporal inclusions of Bacillus thuringiensis. J. Biochem. 2005, 137, 17–25.
- Katayama, H.; Kusaka, Y.; Yokota, H.; Akao, T.; Kojima, M.; Nakamura, O.; Mekada, E.; Mizuki, E. Parasporin-1, a novel cytotoxic protein from Bacillus thuringiensis, induces Ca2+ influx and a sustained elevation of the cytoplasmic Ca2+ concentration in toxin-sensitive cells. J. Biol. Chem. 2007, 282, 7742–7752.
- Hayakawa, T.; Kanagawa, R.; Kotani, Y.; Kimura, M.; Yamagiwa, M.; Yamane, Y.; Takebe, S.; Sakai, H. Parasporin-2Ab, a newly isolated cytotoxic crystal protein from Bacillus thuringiensis. Curr. Microbiol. 2007, 55, 278–283.
- Okumura, S.; Ohba, M.; Mizuki, E.; Crickmore, N.; Côté, J.-C.; Nagamatsu, Y.; Kitada, S.; Sakai, H.; Harata, K.; Shin, T. List of Parasporins. Available online: http://parasporin.fitc.pref.fukuoka.jp/list.html (accessed on 29 November 2021).
- Sanahuja, G.; Banakar, R.; Twyman, R.M.; Capell, T.; Christou, P. Bacillus thuringiensis: A century of research, development and commercial applications. Plant Biotechnol. J. 2011, 9, 283–300.
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Mizuki, E.; Inouye, K. Identification and characterization of a novel cytotoxic protein, parasporin-4, produced by Bacillus thuringiensis A1470 strain. Biotechnol. Annu. Rev. 2008, 14, 225–252.
- Chubicka, T.; Girija, D.; Deepa, K.; Salini, S.; Meera, N.; Raghavamenon, A.C.; Divya, M.K.; Babu, T.D. A parasporin from Bacillus thuringiensis native to Peninsular India induces apoptosis in cancer cells through intrinsic pathway. J. Biosci. 2018, 43, 407–416.
- Nagahama, M.; Hara, H.; Fernandez-Miyakawa, M.; Itohayashi, Y.; Sakurai, J. Oligomerization of Clostridium perfringens ε-toxin is dependent upon membrane fluidity in liposomes. Biochemistry 2006, 45, 296–302.
- Nagamatsu, Y.; Okamura, S.; Saitou, H.; Akao, T.; Mizuki, E. Three cry toxins in two types from bacillus thuringiensis strain M019 preferentially kill human hepatocyte cancer and uterus cervix cancer cells. Biosci. Biotechnol. Biochem. 2010, 74, 494–498.
- Okumura, S.; Saitoh, H.; Ishikawa, T.; Wasano, N.; Yamashita, S.; Kusumoto, K.; Akao, T.; Mizuki, E.; Ohba, M.; Inouye, K. Identification of a novel cytotoxic protein, Cry45Aa, from Bacillus thuringiensis A1470 and its selective cytotoxic activity against various mammalian cell lines. J. Agric. Food. Chem. 2005, 53, 6313–6318.
- Okumura, S.; Koga, H.; Inouye, K.; Mizuki, E. Toxicity of Parasporin-4 and health effects of pro-parasporin-4 diet in mice. Toxins 2014, 6, 2115–2126.
- Nelson, K.L.; Brodsky, R.A.; Buckley, J.T. Channels formed by subnanomolar concentrations of the toxin aerolysin trigger apoptosis of T lymphomas. Cell. Microbiol. 1999, 1, 69–74.
- Maagd, R.A.; Bravo, A.; Berry, C.; Crickmore, N.; Schnepf, H.E. Structure, diversity, and evolution of protein toxins from spore-forming entomopathogenic bacteria. Annu. Rev. Genet. 2003, 37, 409–433.
- Ohba, M.; Mizuki, E.; Uemori, A. Parasporin, a new anticancer protein group from Bacillus thuringiensis. Anticancer Res. 2009, 29, 427–433.
- Balabanova, L.; Golotin, V.; Podvolotskaya, A.; Rasskazov, V. Genetically modified proteins: Functional improvement and chimeragenesis. Bioengineered 2015, 6, 262–274.
- Kim, S.B.; Izumi, H. Functional artificial luciferases as an optical readout for bioassays. Biochem. Biophys. Res. Commun. 2014, 448, 418–423.
- He, L.; Friedman, A.M.; Bailey-Kellogg, C. Algorithms for optimizing cross-overs in DNA shuffling. BMC Bioinform. 2012, 13, S3.
- Wedge, D.C.; Rowe, W.; Kell, D.B.; Knowles, J. In silico modelling of directed evolution: Implications for experimental design and stepwise evolution. J. Theor. Biol. 2009, 257, 131–141.
- Pinzon, E.H.; Sierra, D.A.; Suarez, M.O.; Orduz, S.; Florez, A.M. DNA secondary structure formation by DNA shuffling of the conserved domains of the Cry protein of Bacillus thuringiensis. BMC Biophys. 2017, 10, 1–10.
- Stimple, S.D.; Smith, M.D.; Tessier, P.M. Directed evolution methods for overcoming trade-offs between protein activity and stability. AIChE J. 2020, 66, e16814.
- Basit, N.; Wechsler, H. Prediction of enzyme mutant activity using computational mutagenesis and incremental transduction. Adv. Bioinform. 2011, 2011, 1–9.
- Florez, A.M.; Suarez-Barrera, M.O.; Morales, G.M.; Rivera, K.V.; Orduz, S.; Ochoa, R.; Guerra, D.; Muskus, C. Toxic activity, molecular modeling and docking simulations of Bacillus thuringiensis Cry11 toxin variants obtained via DNA shuffling. Front. Microbiol. 2018, 9, 2461.
- BenFarhat-Touzri, D.; Driss, F.; Jemli, S.; Tounsi, S. Molecular characterization of Cry1D-133 toxin from Bacillus thuringiensis strain HD133 and its toxicity against Spodoptera littoralis. Int. J. Biol. Macromol. 2018, 112, 1–6.
- Sriwimol, W.; Aroonkesorn, A.; Sakdee, S.; Kanchanawarin, C.; Uchihashi, T.; Ando, T.; Angsuthanasombat, C. Potential prepore trimer formation by the Bacillus thuringiensis mosquito-specific toxin: Molecular insights into a critical prerequisite of membrane-bound monomers. J. Biol. Chem. 2015, 290, 20793–20803.
- Pacheco, S.; Gómez, I.; Sánchez, J.; García-Gómez, B.I.; Czajkowsky, D.M.; Zhang, J.; Soberón, M.; Bravo, A. Helix α-3 inter-molecular salt bridges and conformational changes are essential for toxicity of Bacillus thuringiensis 3D-Cry toxin family. Sci. Rep. 2018, 8, 10331.
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802.
- Deist, B.R.; Rausch, M.A.; Fernandez-Luna, M.T.; Adang, M.J.; Bonning, B.C. Bt toxin modification for enhanced efficacy. Toxins 2014, 6, 3005–3027.
More