Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by serge roche and Version 2 by Bruce Ren.
The human kinome is composed of about 50 pseudo-kinases with unclear function, because they are predicted to be catalytically inactive; however, they are shown to play an important role in cancer, similar to active kinases. Understanding how these pseudo-kinases promote tumor formation despite their catalytic inactivity is a great challenge, which may lead to innovative anti-cancer therapies. The PEAK1 and 2 pseudo-kinases have emerged as important components of the protein tyrosine kinase pathway implicated in cancer progression. They can signal using a scaffolding mechanism via a conserved split helical dimerization (SHED) module.
- pseudo-kinase
- oncogene
- cell signaling
- cell growth and migration
- tyrosine kinase
- AKT signaling
1. Introduction
The human kinome includes >50 pseudo-kinases that are predicted to be catalytically inactive due to the lack of important residues required for full enzymatic activity [1]. Their mechanistic role in cell signaling remains unclear, but recent structural analyses suggest a scaffolding or allosteric activity by docking additional kinases for efficient protein phosphorylation [1][2][3][1,2,3]. Moreover, some pseudo-kinases have retained active kinase activity through an unconventional mechanism of protein phosphorylation [1][2][3][1,2,3]. These atypical kinases have gained recent interest because they play important roles as active protein kinases in human cancer. For instance, many pseudo-kinases, such as HER3, TRIBL, and JAK2 (named JH2), are overexpressed or mutated in various cancer types and contribute to tumor progression [1][2][3][1,2,3].
The PEAK pseudo-kinases comprise PEAK1, Pragmin/PEAK2, and chromosome 19 open reading frame 35 (C19orf35)/PEAK3 [4][5][4,5]. They include a variable N-terminal sequence with specific interaction motifs (e.g., SH2 and SH3 binding sites) followed by a shared pseudo-kinase domain. Pragmin/PEAK2 (hereafter PEAK2), the family founder, was originally identified as an effector of the small GTPase Rnd2 to mediate cell contraction [6]. PEAK1 was identified later as a novel cytoskeletal-associated atypical kinase [7]. More recently, using an in-silico sequence–structure analysis, we identified C19orf35, as an additional family member, hence the name PEAK3 [8]. PEAK1/2 are essential components of tyrosine kinase (TK) signaling, leading to cell growth and migration [4][5][4,5]. Mechanistically, through tyrosine phosphorylation they allow the recruitment of important intracellular signaling effectors, such as GRB2 and SHC [7][9][10][11][7,9,10,11]. We and other groups have described PEAK1/2 pro-tumor activity [4][5][4,5]. Specifically, these pseudo-kinases are overexpressed in many epithelial cancer types and contribute to aggressive tumor progression by promoting cancer cell growth and invasion [7][10][11][12][13][14][15][16][17][7,10,11,12,13,14,15,16,17]. Importantly, their aberrant expression in tumors may deregulate cancer cell adhesive properties and enable metastatic progression.
Crystallographic studies revealed an important mechanism by which PEAK1/2 pseudo-kinases may regulate oncogenic pathways [8][18][19][8,18,19]. In addition to a classical protein kinase fold harboring an occluded ATP binding site, three crystal structures revealed an original dimerization domain, i.e., split helical dimerization (SHED) [8][18][19][8,18,19]. This conserved module comprises a long helix N-terminal to the pseudo-kinase domain and three additional helices from the C-terminal extension that form an “XL”-shaped helical bundle. The SHED-based dimerization mechanism regulates PEAK1/2 homo- and hetero-dimerization that might influence PEAK-signaling specificity [20]. However, it is not known whether the SHED domain defines an essential mechanism of PEAK signaling.
PEAK3 was identified as a member of the PEAK family, due to the fact that its peptide sequence predicts a similar degraded ATP-binding site in the TK flanked by the SHED module [8] (Figure 1a). Moreover, PEAK3 self-associates in a SHED-dependent manner [21], but its biological activity is poorly characterized. Recently, Lopez et al. reported that PEAK3 expression can prevent CRKII-dependent membrane ruffling [21], suggesting a negative function in cell migration, dissimilar to PEAK1/2. In this study, we investigated PEAK3’s functional role in cancer. We found that PEAK3 promotes cancer cell growth and migration. Moreover, the SHED module has an important function in these effects through a unique PEAK3/PYK2/AKT signaling cascade. Our findings support the existence of a common SHED-dependent mechanism of PEAK1-3’s oncogenic activity, mediated by the recruitment of a unique set of signaling proteins.
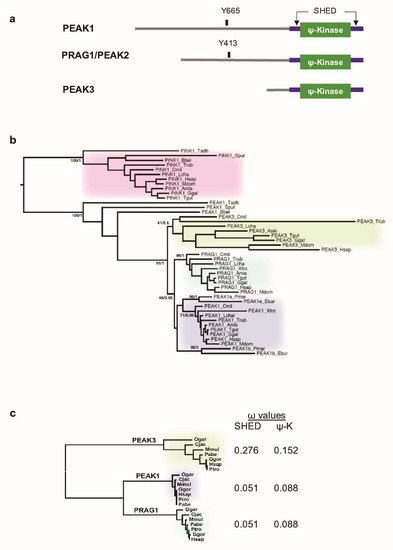
Figure 1. Phylogenetic analysis of PEAK pseudo-kinases. (a) Modular structure of PEAK1-3 pseudo-kinases. The split helical dimerization (SHED) module (made of αN, αJ, αK, and αN helices) (purple), the pseudo-kinase (φ-Kinase) (green), and the main known tyrosine phosphorylation sites are highlighted. (b) Phylogenetic cladogram of PEAK proteins. The tree was deduced from a multiple amino acid sequence alignment of kinase and pseudo-kinase domains and processed by PhyML and MrBayes analysis. PTEN-induced kinases (PINK) were included as an external group to root the tree. Only supports for key phylogenetic nodes are indicated (bootstrap proportion/posterior probability). (c) Analysis of the non-synonymous to silent substitution ratios (ω) of the SHED and pseudo-kinase (ΨK) domains in primate PEAKs. The nucleic sequences of the domains were aligned based on translation and processed with codeml to estimate the ω values. The phylogenetic tree on the left was generated by PhyML analysis of a multiple alignment of the ΨK amino acid sequences.
2. PEAK3 Evolutionary Conservation
Unlike the human genome (three PEAK genes) (Figure 1a), we found a single PEAK gene in the genomes of early metazoan species, from placozoans (Trichoplax adherens) to early chordates (the lancelet Branchiostoma) [21] (Figure 1b) [8]. To better understand the PEAK family ontology, we searched for homologous sequences in key evolutionary species. We identified two PEAK members in cyclostomes (the lamprey, Petromyzon marinus, and the hagfish, Eptatretus burgeri) and three members in cartilaginous fishes (e.g., the elephant shark, Callorhinchus milii), bony fishes (e.g., Takifugu rubripes), and the coelacanth, Latimeria chalumnae. We then extracted the three homologous sequences in additional Tetrapoda species and performed a clustering analysis of MSAs using two probabilistic algorithms (maximum-likelihood and Bayesian). We used the related PTEN-induced kinases (PINK) as the external group to root the tree. The clustering tree globally followed the species phylogeny and showed the presence of a well-supported node leading to three PEAK1-3 clusters in vertebrates (Figure 1b). The branching of the three nodes suggests that a first duplication led to PEAK1/2 and PEAK3, followed by the PEAK1/2 duplication into PEAK1 and PEAK2. This shows that PEAK1-3 emerged at the onset of vertebrates and not in Sauropsida as previously proposed [21]. The presence of only PEAK1-like proteins in cyclostomes suggests that they lost PEAK2 and PEAK3. However, the lower conservation of PEAK3, as indicated by the longer internal branches, may have blurred the branching of the three groups to such an extent that the exact duplication order can no longer be deduced.
To firmly establish the lower conservation of PEAK3 compared with PEAK1/2, we examined the selective constraints exerted on the SHED and pseudo-kinase (ΨK) domains. We calculated the dN/dS ω ratio from a MSA of seven primate species that covered 74 MY of evolution. Neutral selection is associated with ω values close to 1, while values below or above 1 indicate purifying and diversifying selection, respectively. The selection exerted on the SHED and ΨK domains was lower for PEAK3 than for PEAK1 and PEAK2 (Figure 1c), which is in agreement with the difference in branch length. The lowest PEAK3 conservation is also consistent with with the observation that the corresponding gene was lost in several taxa, for instance Myomorpha in rodents. The maintenance of duplicated genes is often associated with neo- or sub-functionalization (i.e., the acquisition of new functions or differential tissue expression, respectively) [22]. Then, we investigated PEAK1–3 mRNA expression using the Human Protein Atlas [23] and found that PEAK1 was ubiquitously expressed, particularly in spleen; PEAK2 expression was more restricted, with the highest expression in cerebellum; and PEAK3 was nearly exclusively expressed in lymphoid tissues (spleen, lymph nodes) and granulocytes. Overall, these data indicate that, although PEAK3 is much less evolutionary conserved than its paralogues PEAK1 and PEAK2, it underwent purifying selection indicative of a physiological function, possibly in blood cells.
3. PEAK3 Overexpression in Acute Myeloid Leukemia
Next, to investigate PEAK3 protein expression in human cells, we generated a polyclonal antibody against full-length recombinant PEAK3. This antibody could detect PEAK3 in lysates from PEAK3-overexpressing U2OS and HeLa cells in western blotting. It also recognized an endogenous protein of about 50 KDa in lysates of THP1 (AML) cells. We confirmed that this protein was PEAK3, because the 50 KDa band disappeared upon siRNA-mediated PEAK3 knockdown. We then used this antibody to assess PEAK3 expression in various human cell lines. We did not detect endogenous PEAK3 in the mesenchymal and epithelia cell lines tested , but only in hematopoietic cell lines, specifically in AML cell lines (Figure 2a). Consistent with this finding, we observed elevated PEAK3 protein expression in 40% of the tested AML patient samples (8/20) (Figure 2b). Analysis of the PEAK3 transcript level from TCGA transcriptomic data (gepia.cancer-pku.cn accessed on 2 January 2021) confirmed that PEAK3 was upregulated in AML (Figure 1c), particularly in the M4 and M5 subtypes. PEAK3 aberrant expression was preferentially associated with specific oncogenic mutations, i.e., DNTM3A mutations, but not with FLT3-TD mutations (Figure 2b). Our findings indicate that PEAK3 is expressed in hematopoietic cells and that its expression can be deregulated in AML, suggesting a pro-tumor function.
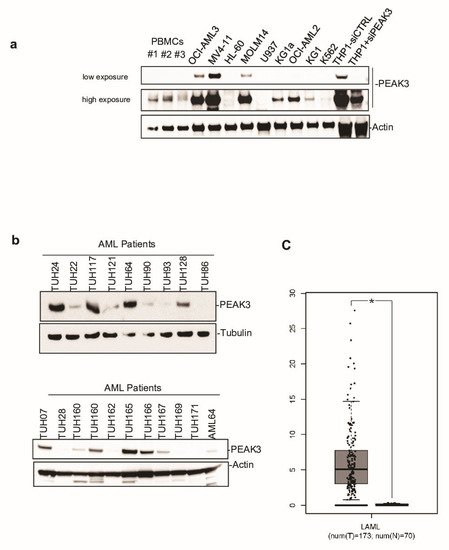
Figure 2. PEAK3 expression in AML. (a) PEAK3 protein expression in the indicated blood cell lines revealed by western blotting with an anti-PEAK3 antibody. Antibody specificity was confirmed using THP1 cells transfected with a siRNA targeting PEAK3. Both low and longer exposure are shown. PBMCs: peripheral blood mononuclear cells (n = 2). (b) PEAK3 expression in AML patient samples. The level of actin or tubulin in also shown (n = 2). (c) PEAK3 upregulation in AML patient samples. The panel shows the relative level of PEAK3 transcripts in 173 AML samples (T) and 70 normal samples (N), (gepia.cancer-pku.cn). * p < 0.05 (Student’s t test).
4. SHED-Dependent Oncogenic Activity of PEAK3
We then investigated whether PEAK3 had pro-tumor functions. PEAK3 overexpression in U2OS cells (Figure 3a) and in THP1 cells (Figure 3c) increased cell growth and invasion in Matrigel-coated Boyden chambers (Figure 3b). These effects were abrogated in both cell lines by overexpression of PEAK3 harboring the A436E mutation in the C-terminal extension of the SHED module (Figure 3a–c), which destabilized its dimerization (Figure 3a) [21]. Similarly, siRNA-mediated PEAK3 silencing in THP1 cells reduced their growth and migration in Boyden chambers coated with an endothelial cell monolayer (Figure 3d). These findings indicate that, in human cancer cells, PEAK3 displays oncogenic activity that requires an intact SHED module. As PEAK1/2 localize at focal adhesions (FAs) [7][13][7,13], we investigated PEAK3 localization by direct fluorescence analysis of U2OS cells that stably express GFP-PEAK3 and demonstrated PEAK3 localization at FAs and partial co-localization with paxillin. We confirmed PEAK3 localization at FAs by indirect immunofluorescence using an anti-HA antibody in U2OS cells that stably express HA-ST-PEAK3. Additionally, both imaging methods showed a strong PEAK3 nuclear localization. Conversely, the PEAK3 A436E mutant did not display FA localization, but was still localized in the nucleus. These observations indicate that PEAK3 localizes at FAs such as PEAK1/2, and that this requires a SHED dimeric module.
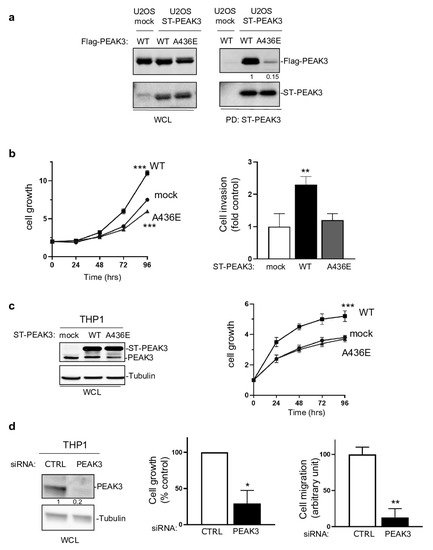
Figure 3. SHED-dependent PEAK3 oncogenic activity. (a) SHED-dependent PEAK3 self-association. Co-precipitation of Srep-Tag-PEAK3 (ST-PEAK3) on Strep-Tactin magnetic beads (PD: pull down) with FLAG-PEAK3 and A436E mutant in U2OS cells stably expressing an empty vector (mock) or ST-PEAK3 that were transfected with indicated FLAG-PEAK3 construct. The level of PEAK3 in whole-cell lysates (WCL) is also shown (n = 2). (b) PEAK3 expression promotes U2OS cell growth in a SHED-dependent manner. Cell growth over time (fold vs. control) (left) and cell invasion in Boyden chambers coated with Matrigel (right) in U2OS cells that express the indicated PEAK3 constructs described in panel A (WT: wild type PEAK3; A436E: PEAK3 with an inactive SHED domain). (c) ST-PEAK3 level (left) and cell growth over time (fold vs. control) (left) in THP1 cells that express the indicated ST-PEAK3 constructs. (d) PEAK3 knockdown (left) reduces THP1 cell growth vs. control cells (cell growth monitored for 3 days) and migration in Boyden chambers coated with an endothelial cell monolayer. Mean ± SD are shown (n = 3); * p < 0.05; ** p < 0.01; *** p < 0.001 (Student’s t test).
5. PEAK3 Activates AKT Signaling
Next, to determine PEAK3 oncogenic signaling, we probed a phospho-kinase antibody array from THP1 cells transfected with a control siRNA or a siRNA targeting PEAK3 (Figure 4a). We found that PEAK3 downregulation affected the activating phosphorylation of adhesive TKs of the Src (SFK) and FAK families and also reduced MAPK and AKT activating phosphorylations (Figure 4a). We confirmed these results using western blotting (Figure 4a, right panel). PEAK3 silencing also reduced phosphorylation of the probed AKT substrates (i.e., GSK-3 alpha/beta, PRAS40, and CREB), suggesting that this pseudo-kinase is a novel upstream regulator of this pathway. Consistently, AKT activity (i.e., pS473-AKT level) increased approximately 2-fold upon PEAK3 overexpression and reduced 3-fold upon PEAK3 knockdown (Figure 4b). We obtained similar results in PEAK3-overexpressing U2OS cells, suggesting that PEAK3 might be a general inducer of AKT signaling (Figure 4c). PEAK3 overexpression induced a modest increase in AKT signaling (i.e., AKT activity and AKT substrate phosphorylation). Conversely, PEAK3 strongly activated AKT in serum-starved conditions (Figure 4c). We did not observe this effect with the PEAK3 A436E mutant, further supporting the essential role of the SHED module in PEAK3 signaling (Figure 4c). PEAK3–AKT signaling was abrogated by pharmacological inhibition of PI3K activity, suggesting that PEAK3 is upstream to PI3K. Collectively, these data indicate that PEAK3 induces PI3K/AKT signaling, even in the absence of growth factors.
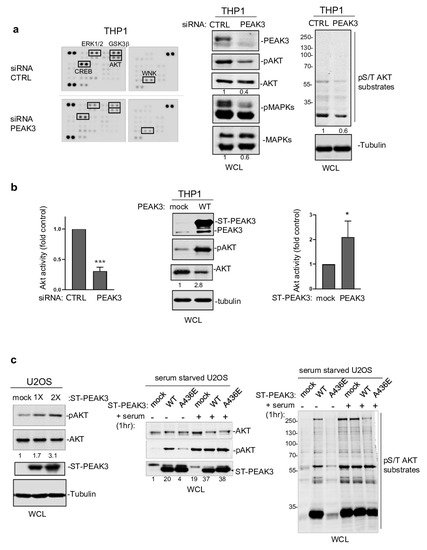
Figure 4. PEAK3 activates AKT signaling in the absence of growth factors. (a) (left) Phospho-kinase array analysis of THP1 cells transfected with siRNA control (CTRL) or siRNA against PEAK3. The phospho-signal reduction in the siRNA PEAK3 condition is highlighted (i.e., pT202/Y204 ERK1/2; pS21/9 GSK3β; pS133 CREB; pS473 AKT; and pT60 WNK). (right) Western blot analysis of MAPK, AKT activity, and signaling (pS/T AKT substrates level). Tubulin level and relative AKT and MAPK activity is shown. (b) (left) Quantification of AKT inhibition upon PEAK3 silencing (mean ± SD; n = 3); *** p < 0.001 (Student’s t test) (bottom). PEAK3 overexpression increases AKT activity in THP1 cells. A representative western blot (middle) and relative quantification of AKT activation (right) (mean ± SD; n = 3); * p < 0.05 (Student’s t test). (c) PEAK3 expression induces AKT signaling in U2OS cells, even in the absence of growth factors. (left) AKT activation and its relative quantification upon transient PEAK3 expression (1×: 1 μg, 2×: 2 μg of PEAK3 construct transfected) (n = 2). SHED-dependent PEAK3 activation of AKT signaling in the absence of growth factors. U2OS cells that stably express WT or A436E PEAK3 were serum-starved for >20 h and stimulated or not with 10% FCS for 1 h. Then, the levels of AKT, phosphorylated AKT (middle), and pS/T AKT substrates were assessed (right) (n = 2).
6. SHED-Dependent PEAK3 Scaffolding Activity
To further elucidate the mechanism by which PEAK3 induces AKT signaling, we performed PEAK3 interactomic analyses of U2OS, HeLa, and THP1 cells that stably express ST-PEAK3 (Figure 5a). Label-free LC-MS/MS analysis of ST-PEAK3 complexes purified on streptavidin beads identified many signaling proteins in the three PEAK3 interactomes, including GRB2, ADP-ribosylation factor (ARF) GTPase-activating proteins ASAP1 and 2, and another member of the PEAK family (i.e., PEAK1 in the HeLa and U2OS cell interactomes and PEAK2 in the THP1 cell interactome). This proteomic analysis also identified many 14-3-3 proteins, which regulate phospho-dependent signaling activity [24] and voltage-dependent anion-selective channel proteins (i.e., VDAC1-3), which regulate cell volume and apoptosis [25]. Additionally, we detected the FAK-like TK PYK2 in the PEAK3 interactome of THP1 cells. We next confirmed some of these findings using western blotting (Figure 5b,c). Although the CRKII cytoskeletal proteins were previously reported as PEAK3 binders [21], we could not detect any endogenous CRKII protein associated with PEAK3 in our proteomic and biochemical analyses (Figure 5c). All these interactions required an intact SHED module (Figure 5c), suggesting that PEAK3 dimerization is essential for interaction with these signaling proteins. As ASAP1/2, VDACs, and PYK2 have not been detected in the previously reported PEAK1 and PEAK2 interactomes [8][20][8,20], PEAK1-3 may recruit a unique set of signaling proteins to induce intracellular signaling.
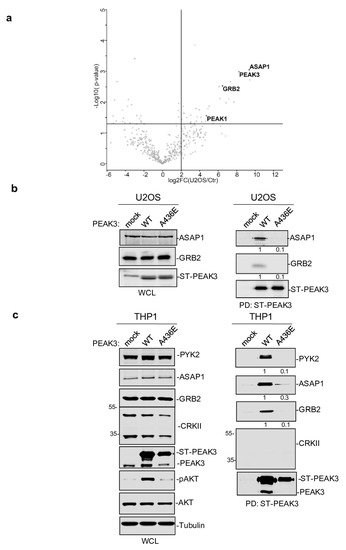
Figure 5. PEAK3 interactomics identified GRB2, ASAP1/2, and PYK2 as SHED-dependent binders. (a) Interactome of PEAK3 in U2OS cells represented as volcano plots of the MS data (n = 3). (b) and (c) Validation by western blot analysis of PEAK3 interaction with the indicated signaling proteins in U2OS (b) and THP1 cells (c) (n = 2). The relative levels of PYK2, ASAP1, and GRB2 associated with ST-PEAK3 WT and AE mutant are shown.
7. PEAK3 Activates PYK2 to Promote AKT Signaling
The fact that PEAK3 interacts with PYK2 suggests that, like PEAK2 [4][8][4,8], PEAK3 may activate specific TKs to mediate phospho-tyrosine signaling. We tested this hypothesis by co-expressing PEAK3 and PYK2 in HEK293T cells. While PEAK3 alone did not clearly affect protein tyrosine phosphorylation, PEAK3 robustly activated ectopic PYK2, as measured on the phosphorylation level of regulatory tyrosine 402 and 881 (Figure 6a,c) [26]. Interestingly, PYK2 activation was accompanied by an increase in cellular protein tyrosine phosphorylation, including a 50 KD and 120 KDa band (Figure 6a). Pull-down experiments next identified PEAK3 and PYK2 some of these tyrosine phosphorylated proteins (Figure 6b). Similar results were obtained in THP1cells where tyrosine phosphorylation of overexpressed PEAK3 was reduced upon acute pharmacological inhibition of PYK2 (PYK2i) (Figure 6c,d). Additionally, the pharmacological inhibition of protein tyrosine phosphatases (pervanadate) largely increased PEAK3 tyrosine phosphorylation (Figure 6c), suggesting the existence of regulatory protein tyrosine phosphatases. Consequently, PEAK3 tyrosine phosphorylation by PYK2 increased its association with GRB2 and ASAP1 (Figure 6b), while PYK2i reduced this effect (Figure 6c,d). ASAP1 was previously identified as a PYK2 substrate [27] and, consistent with our findings, overexpressed PEAK3 increased ASAP1 tyrosine phosphorylation in a PYK2-dependent manner (Figure 6e). Finally, this molecular process required an intact SHED module, suggesting that PEAK3 dimerization is essential for PEAK3 phospho-tyrosine signaling. Overall, these findings support a tyrosine phosphorylation-dependent PEAK3 scaffolding activity, which implicates the TK PYK2.
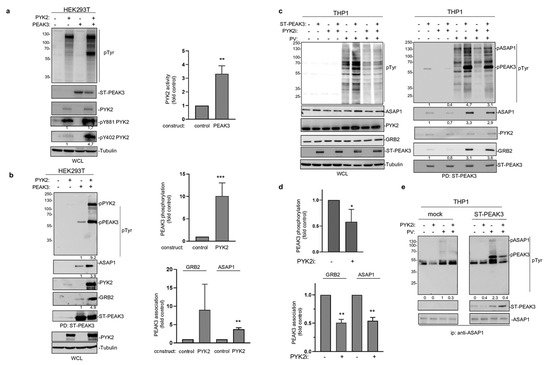
Figure 6. PEAK3 activates PYK2 signaling. (a) PYK2 activation by PEAK3 in HEK293T cells. The levels of protein tyrosine phosphorylation, PEAK3, PYK2, and phosphorylated PYK2 (pPYK2, pTyr402, and pTyr881) were assessed in cells transfected with the indicated constructs for 40 h (left). Relative quantification of PYK2 activation by PEAK3 (mean ± SD; n = 3); ** p < 0.01 (Student’s t test) (right). (b) PEAK3 tyrosine phosphorylation and its association with GRB2 and ASAP1 by PYK2 in HEK293T cells. The level-indicated proteins and their tyrosine phosphorylation in the PEAK3 pull-down were assessed in cells transfected with indicated constructs for 40 h. Relative quantification of PEAK3 tyrosine phosphorylation and its association with GRB2 and ASAP1 (mean ± SD; n = 3); ** p < 0.01, *** p < 0.001 (Student’s t test) (right). (c) PEAK3 tyrosine phosphorylation by PYK2 in THP1 cells. The pull-down of PEAK3 and its associated proteins were assessed in THP1 cells overexpressing PEAK3 and treated or not with pervanadate (PV) (0.1 μM for 15 min) or PYK2i (1 μM for 2h) as indicated. (d) Relative quantification of PEAK3 tyrosine phosphorylation and its association with GRB2 and ASAP1 in THP1 cells (mean ± SD; n = 3); * p < 0.05, ** p < 0.01 (Student’s t test) (right). (e) Increased ASAP1 tyrosine phosphorylation by PEAK3 expression in a PYK2-dependent manner. Immunoprecipitated ASAP1 tyrosine phosphorylation and its association with ST-PEAK3 from indicated THP1 cells treated as described in Figure 6c (n = 2).
This PEAK3 activating mechanism on PYK2 was then confirmed in PEAK3 overexpressing U2OS cells (Figure 7a). Importantly, this molecular effect was further enhanced in serum-starved conditions (Figure 7a), also supporting a growth factor-independent mechanism of PEAK3 activation of PYK2. PEAK3 knockdown reduced PYK2 activity in THP1 cells (Figure 7b), supporting the existence of a similar endogenous PEAK3–PYK2 signaling in leukemic cells. Having demonstrated the existence of PEAK3–AKT and PEAK3–PYK2 signaling, we then asked whether PYK2 would mediate the growth-independent AKT activation by PEAK3. Acute pharmacological inhibition of PYK2 (PYK2i) in U2OS reduced the stimulating effect of PEAK3 on AKT 3-fold, while this inhibitor had no effect on AKT activity in control cells (Figure 7c). Overall, these findings demonstrate the fact that that PEAK3 activates PYK2 as well as the support role of PYK2 activity in PEAK3–AKT signaling.
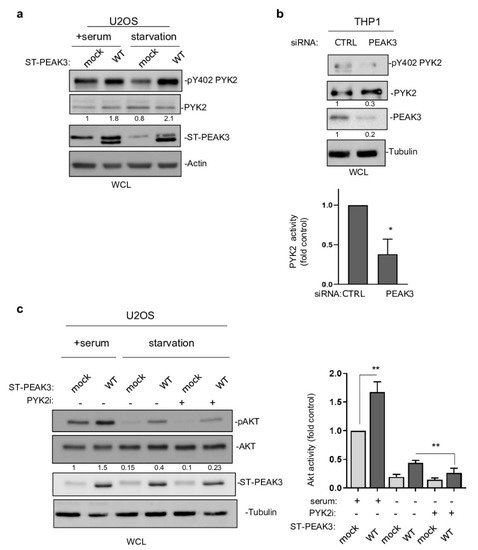
Figure 7. PYK2 activity mediates PEAK3–AKT signaling. (a) PYK2 activation by PEAK3 overexpressing U2OS cells that were serum-starved or not for >20 h. Western blots of the levels of PEAK3, PYK2, and pPYK2 (n = 2). (b) Regulation of PYK2 activity by endogenous PEAK3 in THP1 cells. PYK2 activity (pTyr402 PYK2 level) was measured in cells transfected with the indicated siRNAs. Quantification of PYK2 activity (mean ± SD; n = 3); * p < 0.05 (Student’s t test) (bottom). (c) PYK2 inhibition reduces PEAK3 activation of AKT in the absence of growth factors. U2OS cells that express or do not express PEAK3 were serum starved overnight or not, in the presence of PYK2 inhibitor (PYK2i) (1 μM) or vehicle (DMSO) as indicated. Western blots of the level of PEAK3, AKT, and pAKT (left) and relative quantification of AKT activity (mean ± SD; n = 3); ** p < 0.01 (Student’s t test) (right).