Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ricardo Villa-Bellosta and Version 2 by Jason Zhu.
In vertebrates, plasma and other extracellular fluids are supersaturated with phosphate and calcium, causing a tendency for spontaneous calcium-phosphate precipitation. Therefore, the synthesis of calcium phosphate deposition inhibitors is essential for survival, including pyrophosphate and several proteins.
- vascular calcification
- pyrophosphate
- calcium
- protein
1. Introduction
Matrix Gla protein is a mineral-binding extracellular matrix protein synthesized mainly by vascular smooth muscle cells and chondrocytes, the first protein recognized as an inhibitor of vascular calcification in vitro and in vivo [1][47]. Matrix Gla protein contains several Vitamin K-dependent carboxylation/gamma-carboxyglutamic (Gla) amino acid residues, which are responsible for the high-affinity binding of calcium ions. Notably, matrix Gla protein-deficient mice exhibit spontaneous calcification of the arteries and cartilage, and several studies reported possible associations between plasma Matrix Gla protein and vascular calcification in uremic, diabetic, atherosclerotic, and hypertensive patients. Matrix Gla protein is present in atherosclerotic lesions.
Fetuin-A is a circulating plasma glycoprotein that also has the capacity to bind calcium and has anti-inflammatory properties [2][48]. Notably, Fetuin-A knockout-mice spontaneously develop soft tissue calcification of the heart, vessels, kidney, testis, and skin [3][4][49,50]. However, the relationship between serum fetuin-A levels and vascular calcification remains unclear [5][51].
Osteopontin, a sialic acid-rich glycoprotein first purified from the bone, is a known noncollagenous bone matrix protein that regulates calcification [6][7][7,52]. Like Matrix Gla Protein, osteopontin also regulates calcification during bone development and remodeling. Moreover, osteopontin is also expressed by macrophages, smooth muscle, and endothelial cells in human aortic and coronary atherosclerotic plaques [8][9][53,54]. However, upregulation of osteopontin mRNA levels is not correlated with vascular calcification, suggesting that osteopontin might not be necessary for calcification [5][51]. In addition, several studies suggest that osteopontin is not an endogenous inhibitor of calcification in the aortic wall [10][11][55,56].
On the other hand, extracellular pyrophosphate is the major endogenous physicochemical inhibitor of calcium-phosphate crystal formation and growth, both in vitro and in vivo [12][57]. Extracellular pyrophosphate acts by avidly binding to nascent hydroxyapatite crystals with complete inhibition at micromolar concentration [13][14][15][5,6,11], which is more than 1000-fold less than physiologic calcium or phosphate concentrations. Extracellular pyrophosphate is present at levels sufficient to completely prevent hydroxyapatite formation of physiologic calcium or phosphate concentrations [15][11]. However, loss of extracellular pyrophosphate synthesis or increments of plasmatic phosphate concentration (hyperphosphatemia) lead to vascular calcification due to a lack of inhibitory capacity [16][35]. For example, studies have shown that plasma pyrophosphate is reduced after standard hemodialysis in a mouse model of progeria [17][18][34,58]. Consequently, several studies show that daily injections of exogenous pyrophosphate prevent medial vascular calcification in experimental rat and mice models, including progeria and renal failure [19][20][21][59,60,61]. Notably, several therapeutic strategies that increase endogenous extracellular pyrophosphate synthesis prevent the excessive vascular calcification found in the medial layer of the aortic wall in progeria mice [22][23][62,63]. Therefore, pyrophosphate deficiency [24][64] is a critical risk factor for vascular calcification, suggesting an important role of pyrophosphate homeostasis and extracellular pyrophosphate metabolism in vascular calcification [16][35].
2. Extracelular Pyrophosphate Metabolism
The currently known enzymes and transporters involved in extracellular pyrophosphate metabolism include members of the ecto-nucleotide pyrophosphatase/phosphodiesterase, tissue-nonspecific alkaline phosphatase, ecto-5′-nucleotidase, equilibrative nucleoside transporters, phosphate transporters (NaPi), progressive ankylosis proteins, and pump/channels that release ATP extracellularly, including the multi-drug resistance-associated protein 6 [25][24][26][27][21,64,65,66]. Therefore, understanding the role of enzymes and transporters involved in the extracellular pyrophosphate metabolism could provide potential future therapeutic targets to prevent vascular calcification (see Figure 1).
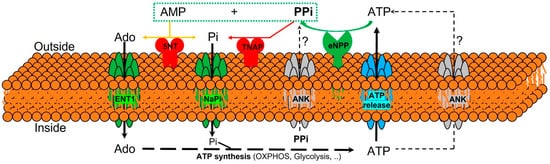
Figure 1. Schematic representation of the ectoenzymes and transporters involved in the extracellular pyrophosphate metabolism. Ectonucleotide pyrophosphatase phosphodiesterase (eNPP) hydrolyze ATP releasing pyrophosphate (PPi) and adenosine-5′-monophosphate (AMP). Pyrophosphate is degraded to phosphate (Pi) by tissue non-specific alkaline phosphatase (TNAP). ATP is released by cells via exocytotic mechanisms and via multiple types of membrane channels, including ABCC6. The progressive ankylosis (ANK) protein can contribute to extracellular pyrophosphate by transporting either ATP or pyrophosphate. Equilibrative nucleoside transporter 1 (ENT1). Sodium-phosphate co-transporter (NaPi). Ecto-5′nucletotidase (5NT). Oxidative phosphorylation pathway (OXPHOS).
Pyrophosphate is mainly produced during the extracellular hydrolysis of ATP [14][28][29][6,67,68]. The major generator of endogenous extracellular pyrophosphate in several tissues, including the aorta, is the enzyme ecto-nucleotide pyrophosphatase/phosphodiesterase (eNPP), which hydrolyzes extracellular ATP to generate pyrophosphate and AMP [28][67]. Three members of the eNPP activity have been found (eNPP1-3); they exist both as membrane proteins, with an extracellular active site, and as soluble proteins in body fluids (also known as PC-1, autotaxin, and B10, respectively) [26][65]. In aorta and vascular smooth muscle cells, eNPP1 is the main source of extracellular pyrophosphate [14][20][29][6,60,68]. Mutations in eNPP1 result in generalized arterial calcification of infancy, characterized by an excessive calcification of the internal elastic lamina of large and medium-sized arteries [28][67]. Moreover, eNPP1-null mice develop ectopic artery calcification [30][69]. Notably, ATP is also a direct inhibitor of calcification [31][70], with a physicochemical mechanism similar to pyrophosphate, bisphosphonates (non-hydrolyzable analogous of pyrophosphate), and polyphosphates [12][32][57,71].
Moreover, pyrophosphate is degraded to phosphate mainly by tissue non-specific alkaline phosphatase (TNAP) in tissues and extracellular fluids. Cells over-expressing TNAP, or the addition of alkaline phosphatase in culture media, is sufficient to cause medial vascular calcification in the aortic ring ex vivo [29][33][68,72]. Notably, TNAP activity is increased in models of medial vascular calcification, such as in uremic rats or in a mouse model of progeria [17][20][34][34,60,73]. Additionally, several studies have shown that phosphatase inhibitors can prevent vascular smooth muscle calcification in vitro and in vivo and that the ablation of phosphatase function produces a loss of skeletal mineralization [35][36][74,75]. TNAP is a non-specific ecto-phosphomonoesterase and a GPI-anchored membrane enzyme, with an extracellular active site and a soluble protein in body fluids. It releases phosphate from various organic compounds, including pyrophosphate [26][36][65,75].
Another enzyme involved in vascular calcification is the membrane-bound ecto-5′nucletotidase (5NT, NT5E, or CD73), which preferentially binds AMP and converts it to adenosine and phosphate. Mutations in ecto-5′nucletotidase induce medial arterial calcification of the lower extremity arteries with peri-articular calcification [37][76]. Like TNAP, ecto-5′nucletotidase is a GPI-anchored enzyme with an extracellular active site and a soluble form cleaved from GPI-anchor. Moreover, like phosphate, adenosine should be recovered from the extracellular space to generate ATP by mitochondria or another metabolic pathway [25][21]. Notably, the first report of a role for adenosine transport in regulating the calcification of soft tissues shows that the loss of the equilibrative nucleoside transporter 1 (ENT1, Slc29a1) in mice could explain the diffuse idiopathic skeletal hyperostosis in humans, characterized by the ectopic calcification of spinal tissues [38][77]. In addition, impaired synthesis of intracellular ATP due to mitochondrial dysfunction has been associated with a reduction in extracellular pyrophosphate concentration, as well as vascular calcification, in a mouse model of premature aging [20][60]. A recent study showed that magnesium treatment improved mitochondrial ATP synthesis and reduced vascular calcification in this mouse model [23][63].
Finally, in 2000 two additional new genes were identified that could play an important role in controlling tissue calcification and arthritis: progressive ankylosis protein and multidrug resistance-associated protein 6. However, the molecular mechanisms remain in part unknown. First, it was reported that mutations in the progressive ankylosis gene cause a severe form of generalized joint calcification and arthritis [39][78]. Loss of progressive ankylosis function causes excessive hydroxyapatite formation in progressive ankylosis gene null mice [39][78]. Overexpression of progressive ankylosis protein in cultured tissue cells increases extracellular pyrophosphate, and cells from the progressive ankylosis protein mutant have a reduction in extracellular pyrophosphate levels [39][78]. In a first study, 7–12 membrane-spanning helices and a central channel for the progressive ankylosis protein [39][78] was proposed. Consequently, it seems as though the progressive ankylosis channel regulates pyrophosphate transport from the cytoplasm to the extracellular milieu; however, additional studies showed that progressive ankylosis protein could be a channel or regulator of adjacent channels which release ATP outside the cells. Notably, in humans, mutations in the channel core of progressive ankylosis protein cause craniometaphyseal dysplasia, a rare skeletal condition of abnormal bone formation characterized by an increased density of craniofacial bones and abnormal modeling of the metaphysis of the tubular bones [40][41][79,80]. Moreover, mutations in the N- and C-terminus of the progressive ankylosis protein cause chondrocalcinosis, a disease of articular cartilage that is radiographically characterized by the deposition of calcium pyrophosphate dihydrate crystals in the joints [42][43][44][81,82,83]. Craniometaphyseal dysplasia is associated with decreased extracellular pyrophosphate levels, whereas chondrocalcinosis is associated with an increase in the amount of pyrophosphate in the extracellular space, which induces the spontaneous formation of calcium pyrophosphate crystals.
Phosphate and pyrophosphate concentration (and, therefore, the phosphate/pyrophosphate ratio), known risk factors for vascular calcification, is strictly controlled by a complex interplay of genes [16][35]. Progressive ankylosis gene and protein could play a key role in this complex process by regulating both eNPP1 and TNAP activities and ATP excretion by different channels. In support of this suggestion, the over-expression of wild-type progressive ankylosis protein results in down-regulation of TNAP activity in chondrogenic cells, and transfection of eNPP1 in osteoblasts enhance extracellular pyrophosphate levels only when wild-type progressive ankylosis protein is present [24][64].
The second-gen reported is multidrug resistance-associated protein 6 (MRP6), also known as ATP-binding cassette sub-family C member 6 (ABCC6) [45][46][84,85]. It was reported that mutations in this gene cause Pseudoxanthoma elasticum, a heritable disorder of connective tissue characterized by calcification of the elastic fibers in skin, arteries, and retina. MRP6/ABCC6 is a member of the superfamily of ABC transporters, composed of several related pumps that can transport various molecules across extra- and intra-cellular membranes, including glutathione-S-conjugates and cyclic nucleotides. This suggests that MPR6/ABCC6 may act as a pump that releases endogenous, low molecular weight inhibitors of calcium phosphate deposits in fluids outside cells, such as ATP or citrate; however, this has not been thoroughly demonstrated.
3. Extracelular Pyrophosphate Metabolism in the Aortic Wall
In the healthy aortic wall, the pyrophosphate synthesis from ATP hydrolysis is several times faster than pyrophosphate hydrolysis [14][29][6,68]. Since vascular smooth muscle cells are the main cell type involved in preventing medial calcification of the aortic wall, the expression and activity of TNAP and eNPP enzymes play a critical role in the prevention of medial vascular calcification [20][22][34][60,62,73]. Notably, both the phosphate-induced aortic and the vascular smooth muscle cells calcification processes, in vitro and ex vivo, respectively, vary depending on the stage of the calcification [14][6]. In the early phase, when calcification is not yet present, eNPP and TNAP activities are increased or decreased, respectively, in vascular smooth muscle cells both in vitro, ex vivo, and in vivo [14][6]. By contrast, in the late phase, when calcification is present and Runx2/Cbfa1 is expressed, hydroxyapatite increases both eNPP1 and TNAP activity, suggesting a compensatory increment in pyrophosphate synthesis in the early phase of phosphate-induced calcification [14][6].
On the other hand, calcification is a very common complication of atherosclerosis that involves aortic smooth muscle cells, monocyte infiltration, and macrophage accumulation within the artery wall [47][4]. In response to a variety of microenvironmental signals, including those found in different regions of atherosclerotic plaques and at distinct stages of atherosclerosis, macrophages polarize, giving rise to a phenotypically heterogeneous cell population with distinct functions [48][86]. Although macrophages display remarkable plasticity and can change their physiology and function in response to environmental cues, atherosclerotic lesion contains cells expressing markers of classical macrophages (M1 macrophages) and alternative macrophages (M2 macrophages), which represent the two major and opposing activities of a wide range of macrophage phenotypes. For example, M1 macrophages promote inflammation, inhibit cell proliferation, and cause tissue damage, whereas M2 macrophages promote cell proliferation and tissue repair [49][87].
Notably, in a macrophage/vascular smooth muscle cell in vitro co-culture system, macrophages enhance the calcifying capacity of vascular smooth muscle cells by inducing phenotypic changes, including matrix mineralization and increment in TNAP activity [50][51][88,89]. Moreover, activators of the vitamin D receptor (calcitriol and paricalcitol) promote calcification in a macrophage/vascular smooth muscle cell co-culture [52][90]. These findings suggest that macrophages could contribute to the calcification of the atherosclerotic plaque in vivo (see Figure 2).

Figure 2. Proposed roles of different macrophage subtypes in calcification of the atheromatous plaque. Classical macrophages (M1 macrophage) induce tissue-nonspecific alkaline phosphatase (TNAP) expression in vascular smooth muscle cells (VSMCs). Moreover, the presence of alternatively macrophages (M2 macrophage) induces ectonucleoside triphosphate diphosphohydrolase 1 (eNPP1) expression in VSMCs. Pi: phosphate; PPi: pyrophosphate.
In a recent study [51][89], the authors show that M2 macrophages also have anti-calcifying properties due predominately to their increased capacity to synthesize extracellular pyrophosphate. M2 macrophages release more ATP and increase pyrophosphate synthesis via increased eNPP1 expression and activity, compared with M1 macrophages. Moreover, a co-culture of vascular smooth muscle cells with M2 macrophages increases eNPP1 expression and activity in vascular smooth muscle cells. In contrast, a co-culture of vascular smooth muscle cells with M1 macrophages increases TNAP expression and activity in vascular smooth muscle cells [51][89].
Finally, a study also shows that hyperphosphatemia can activate macrophages, forming a different and new macrophage type [53][91]. Phosphate-induced macrophages (MPi) express M2 markers and have similar activities to M2 macrophages, including arginine degradation via arginase 1, higher metabolic activity, and increased antioxidant production (see Figure 2). Consequently, as with M2 macrophages, MPi macrophages also possess anti-calcifying properties via increased extracellular pyrophosphate availability. In contrast, calcium-phosphate crystals present in atherosclerotic lesions, including hydroxyapatite, can induce macrophage polarization into M1 macrophages [53][91]. These findings suggest two separate environments and steps are involved during the process of calcification in the atheroma plaque, similar to the two steps also found during medial calcification [14][6]. However, additional deeply studies are necessary [54][92].