In 2000, we prthe co
posed a CaM-based “cork-type” mechan
ism of gap junction chemical gating [112]. This “cork” model envisions a physical obstruction of the cytoplasmic mouth of the channel by a CaM lobe [19][50][112][113], probably combined with conformational changes in the connenexins, caused by Ca
2+-CaM binding to the gating site. The model is based on numerous findings that suggest a direct CaM role in gating; rev. in
[50][51][52][113][50,51,52,113]. Experimental evidence indicates that the chemical/slow gate is a sizable, negatively-charged particle, likely to be a CaM lobe
[69][114][69,114].
There are many reasons why
wresearche
rs think that CaM is the most likely gating candidate. [Ca
2+]
i in the high nM to low μM values activates chemical gating; rev. in
[19][52][19,52]. In view of the fact that the cytoplasmic domains of connexins do not contain sequences able to bind Ca
2+ at such low concentrations, the effect of Ca
2+i on the channel gating is most likely mediated by a CaM-like protein, most likely CaM itself. Indeed, CaM binds to connexins
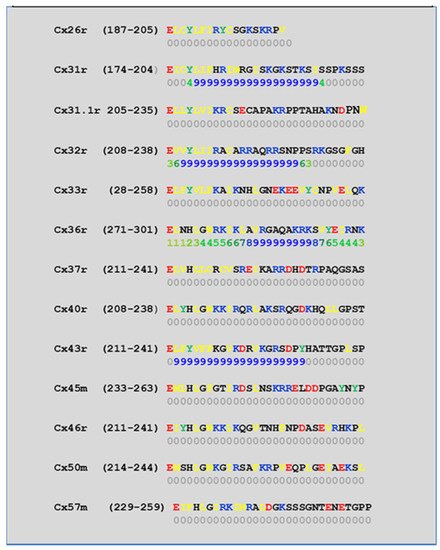
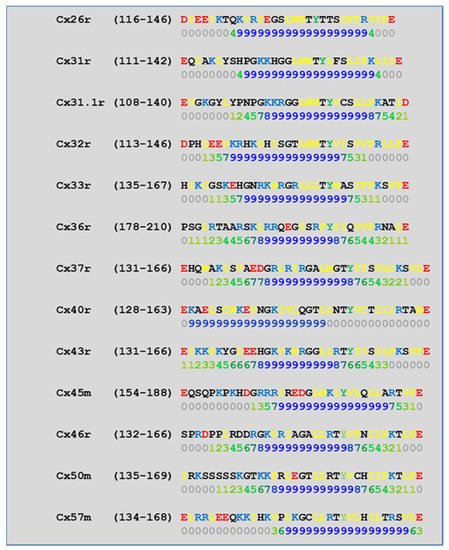
[52][57][58][71][72][77][115][52,57,58,71,72,77,115] which in fact have CaM-binding sites; most of the sites are at the second half of the cytoplasmic loop (CL2), but some are also at the NH
2-terminus (NT-site) and at the NH
2-end of the COOH-terminus (CT1); rev. in
[19][52][75][113][19,52,75,113]. Most relevant for gating are likely to be the CL2 and NT sites
[52][73][74][75][77][108][109][52,73,74,75,77,108,109]. Peptides mimicking the CaM-binding site sequences located at CL2, NT, and CT1 of several connexins bind Ca
2+-CaM with high affinity
[42][52][73][74][75][76][78][79][80][99][103][42,52,73,74,75,76,78,79,80,99,103]. Most important is the binding of CaM to the CL2 domain, which has been experimentally confirmed by Jenny Yang’s team for Cx43
[80], Cx44
[79] Cx50
[78], and Cx45
[77] and by Katalin Török’s team for Cx32, Cx35, Cx45, and Cx57
[73][74][73,74]. CaM and connexins co-localize at gap junctions (
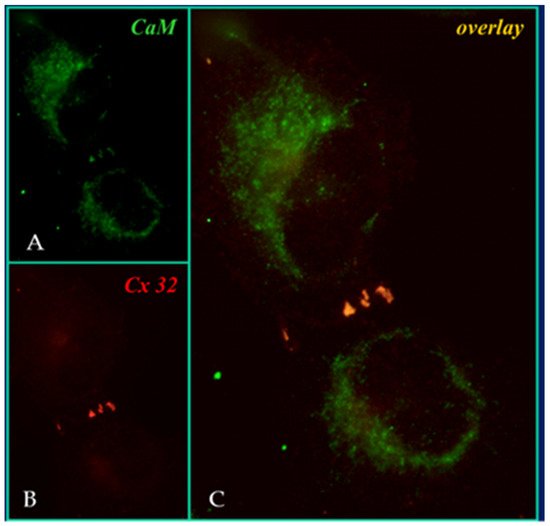
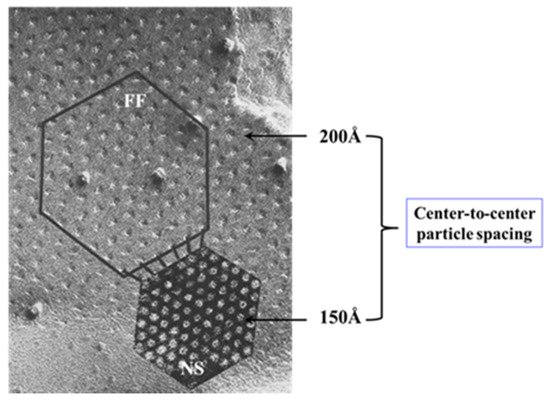
Figure 4) and intracellular sites
[71][72][77][104][116][71,72,77,104,116]. Recently, the direct binding of CaM to Cx45 has been visualized in living cells by Bioluminescence Resonance Energy Transfer (BRET)
[77]; the interaction of CaM and Cx45 was Ca
2+-dependent and prevented by W7; the CL2’s CaM binding site (res. 164–186) was confirmed by a study reporting its high-affinity interaction (K
D = ~5 nM) with a peptide matching the CL2 domain of Cx45’s CL2, tested with a fluorescence-labeled CaM
[77]. On the other hand, however, another study provided evidence for both Ca
2+-dependent and Ca
2+-independent CaM-binding to the CL2 domains of Cx45, Cx32, Cx35, and Cx57
[73][74][73,74]. The Ca
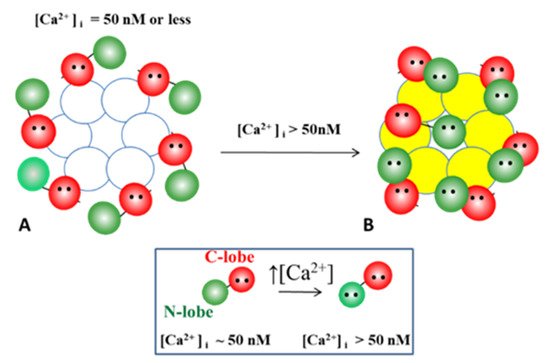
2+-independent binding of CaM to the CL2 domain
[73][74][73,74] confirms earlier data suggesting that the CaM is anchored to the Cxs at normal [Ca
2+]
i (~50 nM)
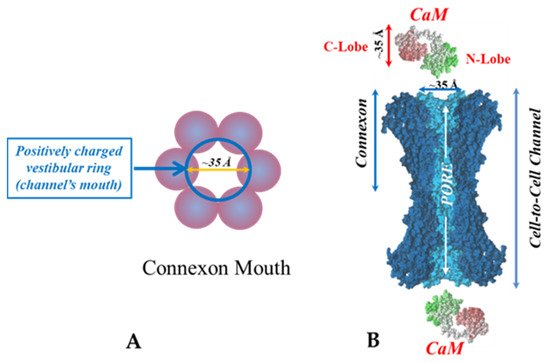
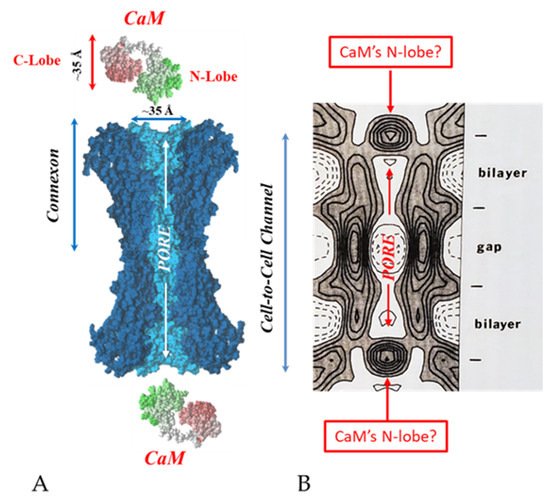
[69][71][72][77][69,71,72,77]. Each of the two negatively-charged CaM lobes is ~25 × 35 Å in size
[91], which is the approximate size of the positively-charged cytoplasmic mouth (vestibule) of the channel
[117][118][119][117,118,119] (
Figure 95). So, a CaM lobe would fit nicely in the mouth (vestibule) of the connexon (
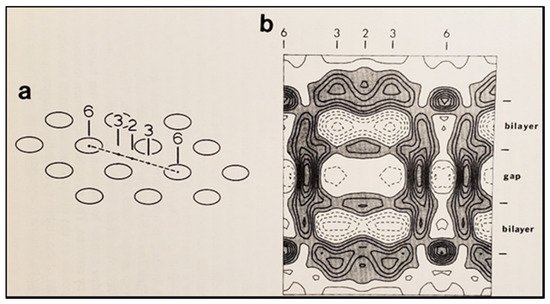
Figure 95B). Evidence from a three-dimensional electron density map of isolated gap junctions, which display crystalline (hexagonal) channel arrays (see in the following), studied by X-ray diffraction, proves that the channels are in a closed state as they are inaccessible to sucrose due to a blocking particle at both channel ends similar in size to a CaM lobe
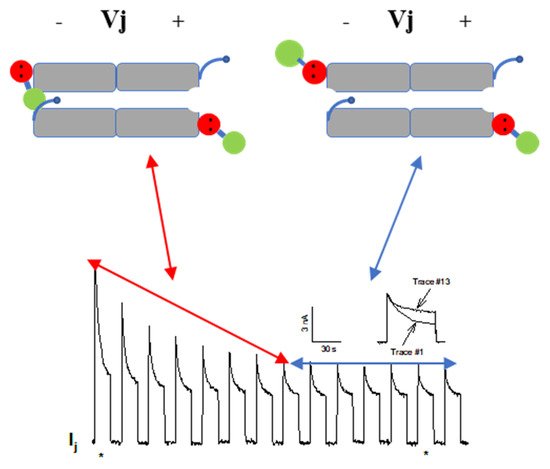
[120][121][122][120,121,122] (see in the following). Significantly, in a double-whole-cell-clamp (single-channel) study the chemical/slow gate opens and closes fully and very slowly (transition time = ~10 ms)
[123] and the open-to-closed channel transitions, and vice versa, often displayed fluctuations
[123]. This further supports the idea that a large particle may transiently flicker in and out of the mouth of the channel before closing the channel completely
[123].
54.1. Ca-CaM-Cork Gating Mechanism
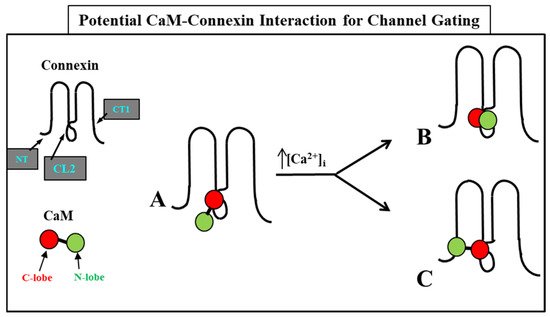
The Ca-CaM-cork gating envisions that a [Ca
2+]
i rise above resting levels (>~50 nM) causes a CaM lobe (most likely the N-lobe) to block the channel’s mouth (vestibule) by electrostatically and hydrophobically interacting with a receptor site located at CL2 or NT (CaM-gating site) (
Figure 106). This interaction is also likely to induce a change in the connexin conformation. The CaM-gating site is likely to be close the channel’s mouth (vestibule)
[19][112][19,112]. At basal [Ca
2+]
i (~50 nM), the CaM is believed to be anchored to each of the six connexins by one of its lobes (most likely the C-lobe), while the other lobe is likely to be free, but unable to access the channel’s mouth at resting [Ca
2+]
i (
Figure 106).
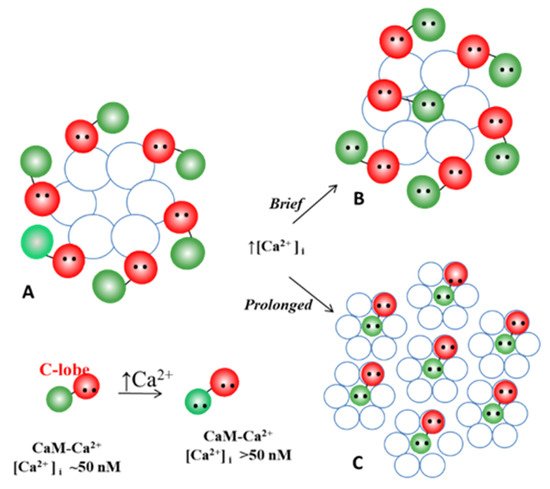
Figure 106. WResearchers believe that CaM is anchored to CL2 by its C-lobe lobe (A). [Ca2+]i > ~50 nM are believed to cause the N-lobe to gate the channel by interacting hydrophobically and electrostatically with CL2 (B) or NT (C) domains of the same connexin or another connexin of the same connexon (trans-domain or trans-subunit interaction, respectively), resulting in pore blockage (cork gating model).
The Ca
2+-affinity constant of the C-lobe’s EF-hand pair is almost one order of magnitude greater than that of the N-lobe’s EF-hand pair (K
D(app) = 5.6 μM and 32 μM for C-lobe and N-lobe, respectively)
[125][126][125,126]. Therefore, the N-lobe most likely interacts with the channel’s gating site (CL2 or NT) only with an increase in [Ca
2+]
i above the resting values (>~50 nM) (
Figure 106).
The Ca-CaM-cork gating is likely to be either reversible or irreversible (Ca-CaM locked gate; see in the following). The former may be activated by a moderate increase in [Ca
2+]
i, while the latter may result from a greater increase and/or a more prolonged [Ca
2+]
i rise.
WResearche
rs believe that the ultrastructural correlate of the “irreversible Ca-CaM locked gate” is reflected by gap junctions displaying “crystalline” (hexagonal) channel arrays
[18][127][128][129][130][18,127,128,129,130] (see in the following).