2.3. Pathology of NeuroAIDS
The pathogenesis of HAND itself is complicated and intricate, and it has been revealed in recent studies that functional alterations in neurons were associated with the pathophysiology of HAND.
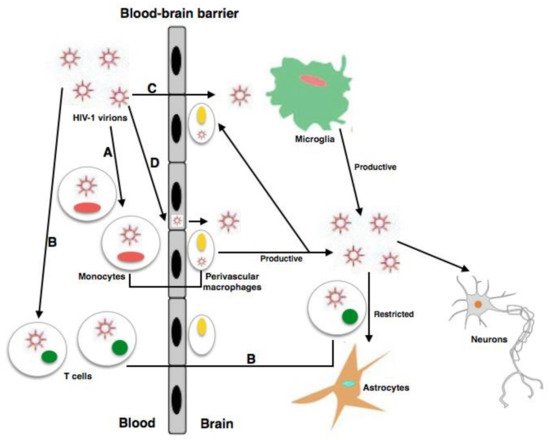
Productive HIV infection occurs in perivascular macrophages, MDMs, and microglia
[20][34]. While it is sufficiently established that neuronal injury and loss are correlated with the evolution of HAND manifestations, there is a paucity of available information on the capability of HIV to infect neurons
[22][23][13,35]. In accordance with the broadly accepted model, in a systemic infection, after at least 1 week, the virus enters the CNS through the BBB, infecting it via a “Trojan horse” pattern
[21][36] (
Figure 1). HIV-1 uses infected lymphocyte cells, CD4 + T cells, and monocyte cells, which later differentiate into macrophages
[23][35], to infiltrate, infect, and activate cells that are in direct contact with perivascular macrophages and astrocytic and microglial cells; subsequently, they transmigrate to the perivascular space of the CNS while evading immune detection
[24][37]. Although astrocytes are sensitive to HIV infection, they do not promote productive infection; conversely, perivascular macrophages and microglia are the only cells in the CNS able to support HIV infection in the brain
[22][21][13,36] (
Figure 1). In the setting of HAND, symptoms are correlated with loss of neurons and cellular damage. The importance of secretion by activated microglial cells, macrophages, and astrocytic cells of small metabolite chemokines, inflammatory cytokines, and neurotoxic viral proteins has previously been demonstrated, and they can cause significant neuronal damage and disrupt the BBB, producing a continuous viral influx
[25][38].
Figure 1. The mechanisms of HIV infection within the CNS. (A, B, C) HIV-1 enters the CNS through diverse pathways: (A) the Trojan horse process through which HIV-1-infected monocytes pass across the BBB and differentiate into perivascular macrophages; (B) the transfer into the CNS of HIV-1-infected CD4+ T cells; (C) entry into brain is possible in a direct way provided their is raised permeability, owing to dysfunctions and/or modified tissue. (D) Microglia, neurons, and astrocytes are the CNS-resident cells vulnerable to HIV-1 infection. Cell activation plays an important role in the release of proinflammatory cytokines and can increase changes in and the permeability of the BBB, thus helping the development of neuro-invasion of HIV and other viruses.
A controversial question concerns the ability of HIV to produce infection in neuronal cells, even at low levels, and a large number of studies, since the 1980s and 1990s, have reported the capability of HIV to produce infection in neuronal cells in the CNS in vivo
[25][38]. Important studies utilizing in situ PCR and immunohistochemistry reported the existence of HIV genetic material and antigens in neuronal cells
[25][38]. Additional research isolating neurons from autopsy brain tissues of PLWH utilizing laser capture microdissection (LCM) demonstrated the presence of HIV pro-viral DNA in neuronal cells by PCR
[26][27][39,40]. Another study used hyperbranched multi-displacement strategies for whole-gene amplification via PCR and analyzed the existence of HIV DNA in neuronal cells from post-mortem brain tissue obtained by LCM
[28][41]. In vitro studies also demonstrated the possibility that HIV-1 could infect human neuronal cells
[29][42]. Nevertheless, authentication of pathologically significant infection of human neurons in vivo remains to be demonstrated. In addition to its existence in the CNS of PLWH, HIV has also been discovered in the CNS and in the developing fetal brain of infected pediatric patients
[30][43].
The theory postulated by the direct model is that infected cells infiltrate the CNS, secreting viral proteins, and produce neuronal impairment through direct connection with neurons
[22][24][13,37]. The indirect theory propounds that neuron injury is arbitrated by the inflammatory response provoked by infected and uninfected glial cells within the CNS against viral infection and HIV proteins released by directly infected cells
[22][24][13,37]. HIV-1 can lead to an inflammatory response produced by the secretion of viral proteins (viral surface glycoprotein 120 (gp120), transactivator of transcription (Tat), and viral protein R (Vpr)), several soluble molecules and cellular products (such as quinolinic and arachidonate acids, adenosine triphosphate (ATP), platelet-activating factor (PAF), excitatory amino acids, superoxide anions, matrix metalloproteases, growth factors, and proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and interleukin-1 (IL-1), C-C Motif Chemokine Ligand 2 (CCL2), and Regulated upon Activation, Normal T cell Expressed and Secreted protein (RANTES))
[22][31][13,44], and glutamate and nitric oxide (NO) radicals secreted by astrocytic cells, causing their cell death
[13][32][33][26,46,47]. There are some molecules that have a neuroprotective effect such as growth factors and β-chemokines, and others that have an adverse neurotoxic role such as excitatory amino acids and other agonists of the N-methyl-D-aspartate glutamate receptor (NMDAR), which also reduce glutamate uptake
[22][31][13,44].
Stimulation of Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasomes in microglia can be produced by the HIV-1 Tat protein as well as raised caspase-1 and IL-1β levels, which intensify and cause exacerbated inflammation
[31][44]. Moreover, Beclin-1-dependent autophagy activation caused by HIV-1 infection releases the p24 viral protein and cytokines that activate the proinflammation response.
In addition, HIV-1 infection produces damage and dysfunction of mitochondria and oxidative events, with the release of reactive oxygen species (ROS), reactive nitrogen species (RNS), and inducible hypoxia factor (HIF)-1 in microglial cells
[31][44].
2.4. HIV Viral Proteins Involved in Neuropathogenesis
The most profoundly examined and possibly most relevant viral protein is the neurotoxic surface protein gp120; moreover, it is implicated as an important factor in the pathogenesis of HAND.
When the neuronal
N-methyl-
D-aspartate glutamate receptors (NMDARs) are activated, the phenomenon known as excitotoxicity occurs due to the excess of glutamate, which causes an excessive calcium flow, the formation of free radicals (NO), mitochondrial damage, and ROS generation, concomitant with lipid peroxidation and caspase activation, which causes damage to neurons, dendritic degeneration, and apoptosis
[31][44].
The gp120 protein causes neuronal injury by activation of
N-methyl-
D-aspartate (NMDA)-coupled NMDARs, resulting in unusually high calcium flow, while Tat phosphorylates NMDARs, which potentiates glutamate excitotoxicity
[31][44]. Both Tat and Vpr have also been shown to be involved in neuronal impairment, with neuronal cell death related to Tat
[34][49]. In addition, gp120 promotes oxidative events and dysfunction, generation of ROS, and raised cerebral endothelial permeability
[35][50].
The gp120 protein has many functions, and among them, it has the ability to produce the release of inflammatory cytokines and neurotoxic components
[36][51] that generate neuron damage in the CNS and some downregulation of tight junction proteins
[37][52]. The entry of the virus and the induction of an effective infection in the cells are promoted by the action of different elements such as the presence of CD4, the chemokine receptor CXC 4 (CXCR4), and the chemokine receptors 3 and 5 (CCR3 and CCR5) mediated by protein kinase C (PKC) coreceptors, in addition to processes such as intracellular calcium secretion
[37][52]. The apoptotic processes are influenced by gp120, which causes dysregulation of calcium homoeostasis, stimulation of oxidative stress, and activation of the proapoptotic transcription factor p53
[38][53].
Previous studies have revealed a destructive and crucial function of HIV-1 Tat in the development and progression of HIV-1 infection
[39][54], and when the disease is manifested, it is one of the first HIV proteins to be expressed. Several studies have revealed that Tat contributes to neuronal impairment in diverse ways. Tat crosses the BBB because of its ability to modify the expression pattern of proteins critical for the integrity of the endothelial tight junctions
[37][52], and to promote leukocyte infiltration and invasion
[38][53].
In previous studies, Tat was revealed to have cytotoxic and proinflammatory effects that stimulate microglia; synthesize proteins, molecules, and other factors such as monocyte chemoattractant protein type 1 (MCP-1), adhesion molecules of the CAM protein family (V-CAM 1 and I-CAM1), PAF
[39][54], and free radicals; and interfere with molecular mechanisms controlling cyclic adenosine monophosphate (AMP) levels, intracellular calcium concentration, and ion channel expression
[39][54]. The exposure of human astrocytoma cells to the HIV-1 Tat recombinant protein produced high levels of apoptosis compared to untreated cells
[39][54]. In accordance with this finding, cerebral injection of Tat within mice provoked raised Ca2+ v1.2 channels, producing astrogliosis in the cortical region and successive death of cortical neurons, microglial cells, and MDMs
[40][55].
HIV-1 Vpr is released from cells infected by HIV-1. This protein is involved in the apoptosis process, presumably through the production of IL1- and interleukin 8 (IL-8), which promote the release of neurotoxins such as matrix metalloproteinases and induce proteins involved in the cell cycle and proapoptosis
[41][58]. This protein activates the release of proinflammatory cytokines, such as TNF-α, IL-1, and IL-8 in MDMs, and presumably can act on the mitogen-activated protein kinase (MAPK) pathway
[41][58].
HIV-1 negative regulatory factor (Nef) is a flexible, multifunctional protein with several cellular targets that is necessary for the pathogenicity of the virus
[42][61]. This protein provokes astroglial cell activation and astrogliosis
[43][62]. Nef favors lysosome permeabilization, stimulating the release of enzymes
[44][63] and the successive cell death of microvascular endothelial cells (MVECs)
[45][64].
The full length of Tat has a total length of 86–104 amino acids, and peptide analysis of different overlapping lengths did not yield neurotoxic processes in cultures of primary neurons. After this study, it was investigated whether Tat was toxic to fetal neurons cultures through a calcium-dependent process or by raising oxidative stress. Previous studies utilized direct intra-striatal injections of Tat that result in increased carbonyl formation
[46][67]. Increased gliosis, cellular injury, and neuronal death have been associated with an increase in apoptosis
[47][68]. Alteration of calcium homeostasis, activation of TNF-α, nuclear factor κappa-light-chain-enhancer of activated B cells (NF-κB), and glutamate receptors, and stimulation of NO production are the basis of Tat-mediated neurotoxicity. Similar to Tat, gp120 has been found to provoke neurotoxicity via different pathways. In both in vivo and in vitro experiments, gp120 exposure has been demonstrated to provoke cell death
[48][69]. NMDA decreases gp120-induced toxicity. Stimulation and activation of the NO synthesis pathways have also been shown following the administration of gp120
[48][69].
The Tat protein interacts with the NMDA receptor that produces high levels of Ca2+/calmodulin-dependent protein kinase II, which reacts with glycogen synthase kinase 3β and causes the death of oligodendrocytes, a reduction in myelin-like membranes in mature oligodendrocytes, and HIV-1 neuropathology
[48][69].
2.5. Inflammation and Role of Mononuclear Phagocytes in HAND
In addition to the direct toxicity of HIV proteins, mononuclear phagocytes, including perivascular macrophages and resident microglial cells, have an important impact on the evolution of the most severe level of HAND: HAD.
Inflammation has a crucial function in the production of phenomena that provoke neurodegeneration in this infection. The entry of HIV into the CNS occurs via the BBB, which is carried out by circulating monocytes in response to the chemotactic signals produced within the parenchyma, and the monocytes are responsible for the stability of the disease in perivascular macrophages of the CNS
[28][41], microglia
[20][34], and astrocytes
[49][72].
It has been reported that an important component of the immunoproteasome, TRIM5α, was independently silenced utilizing siRNA, and the impact on IFNα-induced viral elimination was examined. The useful inter-dependence of the IFNα-activated anti-HIV-1 phenotypes of TRIM5α in humans and the immunoproteasome was established by the reduction in the levels of IFNα elimination that was shown following PA28A silencing in cells that lacked TRIM5α
[50][75]. In spite of the existence of sporadic data of human TRIM5α that affects this infection either by elimination of certain HLA-associated cytotoxic T lymphocyte (CTL) escape mutant viruses or by producing autophagy in Langerhans cells, these data show non-strain-specific inhibition of HIV-1 infection by human TRIM5α. Principally, this study reported that TRIM5α is functional in CD4+ T cells and is dependent on IFNα and stimulation of the immunoproteasome. The IFN levels are high during the acute and chronic phases of natural infection with HIV-1, and it can be supposed that TRIM5α is partly responsible for the HIV-1 immune control in patients, a result with important associations between positive clinical results and raised TRIM5α expression or specific TRIM5α alleles
[50][75].
Additionally, an interesting topic concerns the possible effects of HIV-1 inhibitors on possible immunosurveillance, which depend on the particular role of proteasome and CTL epitope generation as well as on the relative improvements in the CTL response as an antagonist to the recovery of CD4+ lymphocyte cells and antibody production
[51][76] in immunosurveillance. Antiviral drugs with proteasome inhibitory capacity, such as ritonavir/saquinavir, are able to modulate the presentation of Ags to CTLs and may perhaps be exploited further to find new strategies for treatment of autoimmune disease, chronic immunopathologies, or disease caused by transplantation reactions.
Morbidity and mortality in HIV infection are mainly due to microbial translocation, probably due to the persistent inflammation that it produces and maintains
[52][53][78,79]. The relationships between disease progression, microbial translocation, and mortality do not depend on viral inhibition by ART in PLWH
[54][80]. Different studies have shown direct correlations between plasma levels of LPS (lipopolysaccharide from the surface of Gram-negative bacterial–microbial translocation products) in residual viremia of PLWH, stimulation of CD38+HLA-DR+ CD8+ T cells, and stimulation of monocytes, interferon-sensitive genes such as MxA, and proinflammatory cytokines such as IFN-α, IL-6, and TNFα.
LPS levels and/or bacterial DNA levels have a direct association with other biomarkers of microbial translocation and innate immune activation, such as soluble CD14 (released by monocytes of bacterial stimulation), LPS-binding protein (LBP), and endotoxin. There are many inflammatory events that occur during HIV infection (virus replication, opportunistic infections, etc.). Studies in which no association with HIV was explored showed that microbial translocation and inflammation are associated. In idiopathic CD4 lymphocytopenia (ICL), LPS is found at high levels and is related to the proliferation of CD4 + T cells; in the colon in uninfected pig-tailed macaques, LPS levels were related to the interferon-responsive MxA gene in the GI tract
[54][80], which shows that microbial products can directly activate inflammatory responses.
According to a recent study, increased neuropathy can occur due to control of viral replication in the brain and the loss of immune stimulation by the gut microbiota. Therefore, the loss of protective resident microbes can lead to CNS dysfunction
[53][79].
LPSs that are sufficient to prime microglial cells for antigens presented to efficiently eliminate the virus have been implicated in CNS maturation
[53][54][79,80]. Toll-like receptors (TLRs), expressed by microglial cells, are thought to act only during infection. It has been shown that the microbiota regulates microglial cell function through TLR-4, which prepares these cells to attack an infection. Microglial cells evolve early in embryogenesis from the progenitors of the yolk sac; however, unlike macrophages, microglial cells live long, without any support from circulating blood cells
[53][79]. In addition, Toll Like Receptor 4 (TLR4) exists in the intestinal microbial products that are circulating in the blood and could thus reach the brain
[55][81]. Evidence exists showing that the microglial cell signaling of TLR4 causes stimulation of the microglial cells, and that signals from the gut microbiota can be transferred to the brain from the enteric nervous system
[50][75]. Actually, it has been reported that enteroendocrine cells have neuropods (axon-like basal structures that contain neurofilaments, which are typical structural proteins of axons) that are connected to neurons and are capable of transferring signals to the brain
[55][81]. Brown et al. administered LPS orally, and this administration blocked effects of gastrointestinal exposure, but oronasal–pharyngeal and pneumonic exposure may occur as well. In relation to this topic, dissimilarities detected between feeding mice LPS alone and in combination with a TLR1/2 ligand (Pam3CysK4) indicate that further investigations are necessary to study the interaction between multiple TLR ligands on microglial cell function and response to infection. Furthermore, in this study, it could not be completely ruled out that there is a contribution from cells of gut-resident CX3CR1 to this phenotype or other migrating DC cells
[56][82]. Important studies have indicated that cells of the gut could reach the CNS, and that there is a population of long-lived CX3CR1 cells in the gut
[57][83]. Other studies postulated that disruption of the gut epithelial barrier may allow unregulated translocation of gut microbes into the lamina propria. Thus, factors of bacteria can penetrate the gut-associated lymphoid tissues (GALT) and lumen of the blood, where they communicate with various immune cells and can trigger effector-type T cell differentiation
[58][84]. Regulatory T cells that survey the GALT, blood, and CSF and modifications to the local microbiome can promote T cell brain penetration. Factors of bacteria could upregulate inflammatory cytokine levels, alter BBB integrity, and produce inflammation
[54][80]. Additionally, factors of bacteria can produce LPSs and can regulate endothelial TLRs to produce brain inflammation and infection within the CNS
[58][59][84,85].
The clinical conclusions of these findings reveal that immunostimulatory LPSs are derived from the microbiota, and the structure of the microbiota and the immune stimulatory products being released by the microbiota likely dictate the maturation of microglial cells in the brain. In addition, fatty acids that can be generated by the microbiota are also able to bind TLR4 and might establish other agonists
[60][61][86,87]. As microglial cells can modify the role of neuron cells, these data propose that microbial control of microglial cells could affect the activity and duty of neurons
[62][63][88,89].