Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 1 by Matthias Bosman.
The anthracycline doxorubicin (DOX) is one of the most efficacious chemotherapeutic drugs for the treatment of a wide array of cancers. However, DOX treatment has been associated with development of cardiotoxicity and eventually heart failure, which limits its clinical use. While DOX-induced cardiotoxicity has been extensively investigated (reviewed elsewhere), the toxic effects of DOX on the vasculature have been less considered. Epidemiological studies point towards accelerated vascular ageing in childhood cancer survivors, as evidenced by a higher incidence of atherosclerosis and hypertension. Moreover, several studies have demonstrated that DOX increases arterial stiffness in cancer patients, both during and after treatment.
- doxorubicin
- cardio-oncology
- arterial stiffness
- endothelial dysfunction
- vascular smooth muscle cell contraction
- non-selective cation channel
1. Introduction
Previously, it was shown that doxorubicin (DOX) impairs flow-mediated vasodilation in the brachial artery in patients shortly after DOX treatment, often referred to as endothelial dysfunction [18][1]. In addition, ourthe research group demonstrated that 2 weeks of DOX administration to mice (4 mg/kg/week) acutely impaired endothelium-dependent vasodilation and consequently increased vascular tone, thereby actively augmenting arterial stiffness [19][2]. We, and oIthers, identified endothelial cell loss and reduced endothelial nitric oxide synthase (eNOS) levels as potential mechanisms contributing to impaired endothelial function after DOX treatment [19,20][2][3]. Other studies have reported that DOX may cause uncoupling of eNOS, which is characterised by increased superoxide formation instead of nitric oxide production, thereby contributing to reactive oxygen species (ROS) generation [21,22][4][5].
Apart from the widely accepted toxic impact of DOX on endothelial cell (EC) function, DOX also affects vascular smooth muscle cell (VSMC) function [23,24,25][6][7][8]. There have been conflicting reports about the role of DOX in the alteration of VSMC reactivity. Gibson et al. reported decreased VSMC contraction in rat thoracic aorta rings 24 h after DOX treatment (15 mg/kg) [23][6]. Similarly, Olukman et al. demonstrated that DOX (20 mg/kg) attenuated VSMC contraction in rat aortic segments 1 week after administration [24][7]. Conversely, Shen et al. described increased VSMC contraction in mouse aortic rings after ex vivo DOX treatment (100 μM), which was attributable to increased Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry in VSMCs [25][8]. In ourthe previous study, we did notthere is no observeation of any direct effects of 2 weeks DOX treatment on VSMC contraction [19][2]. Hence, the exact effects of DOX on modulation of VSMC function remain unclear and may be dependent upon the dose or treatment protocol.
2. A Single Dose of DOX Impairs VSMC Contraction and Endothelium-Dependent Vasodilation
There was a decrease in phenylephrine (PE)-induced contraction 16 h after in vivo DOX administration (Figure 1A). The PE-elicited contraction response, which comprises an initial transient and a subsequent tonic phase [26][9], was further investigated. DOX reduced the tonic but not the transient contraction in aortic segments after addition of 2 μM PE (Figure 1B).
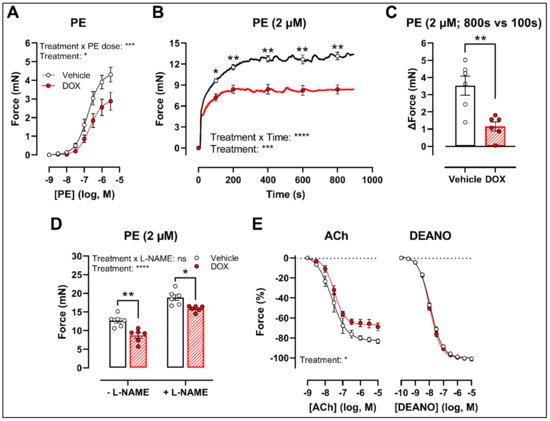
Figure 1. VSMC contraction and relaxation in aortic segments, 16 h after in vivo DOX administration. PE-induced contraction was decreased in aortic segments after in vivo DOX treatment (A). The PE-elicited contraction response was reduced after DOX treatment, which suggests perturbed tonic contraction (B). The increase in force between 800 s and 100 s was lower in the DOX group, indicating that DOX impairs Ca2+ influx (C). The DOX-induced reduction in contraction persisted under L-NAME conditions (D). DOX impaired ACh-induced relaxation, but relaxation of aortic segments in response to DEANO did not change (E). For (A,B,E), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. For (C), unpaired t-test. For (D), two-way ANOVA with Tukey’s multiple comparisons test. *, **, ***, **** p < 0.05, 0.01, 0.001, 0.0001; n = 6 for each group.
Since the tonic contraction phase is dependent on Ca2+ influx [26][9], determining the increase in contraction force (Δ) at different time points offers an estimate of the effectiveness of Ca2+ influx in VSMCs. Here, weit calculated the increase in force (Δ) by subtracting the force at 800 s from the force at 100 s during the PE-elicited contraction response. This increase in contraction force (Δ) was lower in the DOX-treated group compared with the vehicle-treated group (Figure 1C), which suggests that DOX impairs Ca2+ influx.
The contraction force remained lower in the DOX-treated group in the presence of Nω-nitro-L-arginine methyl ester (L-NAME) (Figure 1D). Of note, the PE contraction in panel D is the maximal amplitude of panel B, and these contraction amplitudes are higher than in panel A due to a decrease of basal nitric oxide (NO) over time [27][10]. Further, impaired acetylcholine (ACh)-induced endothelium-dependent relaxation was observed after DOX, while diethylamine NONOate (DEANO)-induced endothelium-independent relaxation was not affected (Figure 1E). These results suggest that DOX decreases VSMC contraction through a VSMC-specific mechanism.
3. Ex Vivo Incubation of Aortic Rings with DOX Leads to Reduced VSMC Contraction and Impaired Endothelium-Dependent Relaxation
To develop a drug testing platform for studying acute effects of DOX and to corroborate the in vivo DOX treatment findings, isolated aortic segments were isolated from mice and subsequently treated with DOX (1 μM) ex vivo for 16 h. Similar results were obtained under these experimental conditions. More specifically, PE-induced contraction was reduced after DOX treatment in a PE concentration response curve (Figure 2A) and contraction-over-time curve (Figure 2B). The increase in force (Δ) was lower for DOX-treated aortic segments compared with the vehicle group (Figure 2C). Under L-NAME conditions, the contraction remained decreased in the DOX group (Figure 2D), and there was impaired ACh-induced relaxation and unaltered DEANO-induced relaxation (Figure 2E).
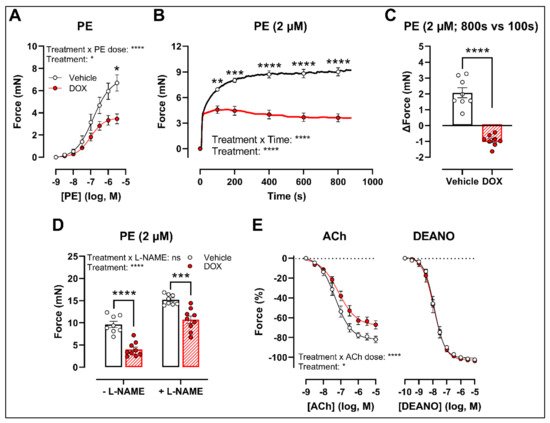
Figure 2. VSMC contraction and relaxation in aortic segments after 16 h of ex vivo DOX treatment. Following 16 h of ex vivo DOX treatment, VSMC contraction was decreased in response to PE (A,B). In the DOX-treated aortic segments, there was a lower increase in force between 800 s and 100 s (C). The DOX-induced decrease in contraction was still observed in the presence of L-NAME (D). ACh-induced relaxation, but not DEANO-induced relaxation, was impaired in response to DOX (E). For (A,B,E), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. For (C), unpaired t-test. For (D), two-way ANOVA with Tukey’s multiple comparisons test. *, **, ***, **** p < 0.05, 0.01, 0.001, 0.0001; n = 8 for vehicle group and n = 9 for DOX group.
4. DOX Does Not Modulate Ca2+ Release from the Sarcoplasmic Reticulum or Ca2+ Influx through Voltage-Gated Calcium Channels
To further investigate the molecular mechanisms of DOX-reduced VSMC contraction, the PE contraction (2 μM) was repeated under 0 mM Ca2+ Krebs Ringer and CaCl2 (3.5 mM Ca2+) conditions to specifically evaluate the transient contraction phase, caused by Ca2+ release from the sarcoplasmic reticulum, and the tonic contraction phase, characterised by VGCC- and NSCC-mediated Ca2+ entry, respectively. To avoid the possible influence of NO, experiments were performed under L-NAME conditions. Contraction mediated by Ca2+ release from the sarcoplasmic reticulum and decline in contraction due to Ca2+ uptake and Ca2+ efflux were not altered in response to DOX (Figure 3A). Upon addition of 3.5 mM Ca2+, thereby promoting Ca2+ entry through voltage-gated calcium channels (VGCCs) and non-selective cation channels (NSCCs), the resulting VSMC contraction was lower in the DOX-treated group compared with the vehicle group (Figure 3B). Subsequent addition of the VGCC blocker diltiazem (35 μM) revealed similar relaxation between the vehicle- and DOX-treated aortic segments (Figure 3C). VSMC contraction, elicited by different doses of K+ (10, 20, 30, 40 and 50 mM), was not altered after DOX treatment (Figure 3D). Moreover, incubation of aortic segments with the VGCC deactivator levcromakalim (1 μM), prior to addition of PE, resulted again in decreased tonic contraction in the DOX-treated group compared with the vehicle group (Figure 3E). These results suggest that DOX does not decrease VSMC contraction through molecular mechanisms responsible for intracellular Ca2+ release or Ca2+ uptake, Ca2+ efflux and VGCC-mediated Ca2+ entry.
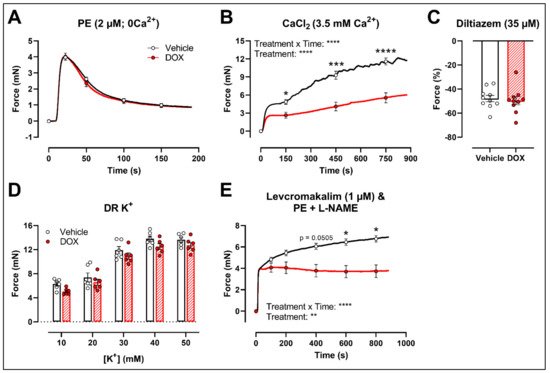
Figure 3. Contraction of aortic segments mediated by Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry through VGCCs and NSCCs after 16 h of ex vivo DOX treatment. (A) Contraction through Ca2+ release from the sarcoplasmic reticulum in response to PE (2 μM) in the absence of extracellular Ca2+ (0 mM Ca2+). (B) Addition of 3.5 mM Ca2+ after 0 mM Ca2+ PE-elicited contraction. (C) Addition of 35 μM diltiazem following stable CaCl2-mediated contraction. (D) Dose–response of K+-elicited contraction (10, 20, 30, 40 and 50 mM). (E) PE-induced contraction (with L-NAME) under levcromakalim (1 μM). DOX did not change contraction through Ca2+ release from the sarcoplasmic reticulum, nor Ca2+ uptake and Ca2+ efflux after 16 h of ex vivo DOX treatment (A), but VSMC contraction was decreased in the DOX-treated group after CaCl2 addition (B). There was no difference in VSMC relaxation between the groups after blocking of VGCCs with diltiazem (C), nor in the contraction magnitude in response to different K+ doses (D). DOX decreased PE-induced contraction (with L-NAME) in the presence of levcromakalim (E). For (A,B,D,E), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. For (C), unpaired t-test. *, ***, **** p < 0.05, 0.001, 0.0001; p > 0.05 in (A,C,D); (A–C) n = 10 in each group; (D,E) n = 6 in each group.
Another possibility is that DOX affects NSCC-mediated contraction instead. Accordingly, further experiments were performed using several pharmacological agents blocking a variety of NSCCs, such as tranilast, SKF-96365, 2-aminoethoxydiphenyl borinate (2-APB) and 9-phenantrol. To avoid any possible inhibitory effect of NO on VGCCs and NSCCs, the following experiments were performed under L-NAME conditions.
Similar to the decrease in PE-elicited contraction (with L-NAME) under levcromakalim, DOX reduced the PE-induced VSMC contraction (2 μM) in aortic segments pre-incubated with diltiazem (35 μM) (Figure 4A). Subsequent addition of tranilast (100 μM), SKF-96365 (10 μM), 2-APB (20 μM) or 9-phenantrol (1 μM) resulted in a decline in VSMC contraction, which did not differ in absolute values (Figure 4B) but was higher in the DOX-treated group when expressed relative to its preceding contraction (Figure 4C).
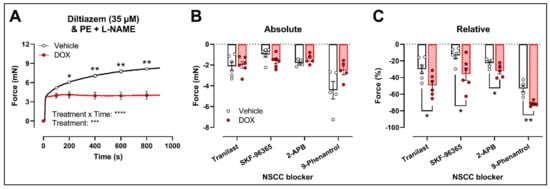
Figure 4. Evaluation of the involvement of members of the NSCC family in decreased PE-induced contraction in aortic segments after 16 h of ex vivo DOX treatment. (A) PE-induced contraction (2 μM, with L-NAME) under diltiazem (35 μM). (B,C) Absolute and relative decline in PE-induced contraction (with L-NAME and diltiazem) under different NSCC-blocker conditions. DOX decreased PE-induced contraction (with L-NAME) in the presence of diltiazem (A). In addition, the DOX-treated group did not display a significant change in VSMC contraction in absolute values (B) but showed a greater decline in VSMC contraction when expressed relative to its preceding contraction magnitude under tranilast (100 μM), SKF-96365 (10 μM), 2-APB (20 μM) and 9-phenantrol (1 μM) (C). For (A), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. For (B,C), unpaired t-test for each NSCC blocking condition. *, **, ***, **** p < 0.05, 0.01, 0.001, 0.0001; p > 0.05 in (B); n = 6 in each group.
Finally, to evaluate whether the decrease in VSMC contraction after DOX treatment was specific for the PE-elicited response, additional contraction agonists were used. DOX-treated aortic segments showed decreased VSMC contraction in response to 0.25 μM endothelin-1 (Figure 5A) but not after 2 μM angiotensin II (Figure 5B), 2 μM serotonin (Figure 5C) and 50 mM K+ (Figure 5D) stimulation.
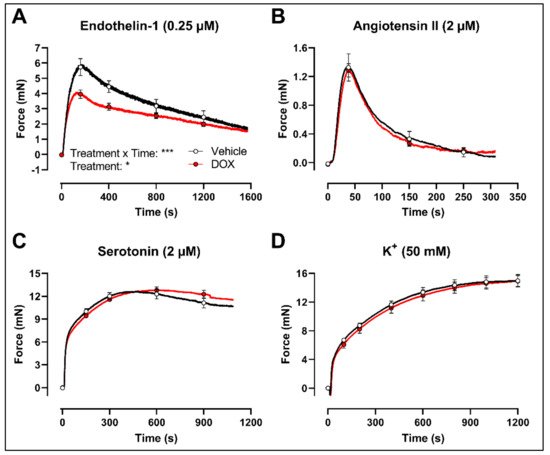
Figure 5. Contraction of aortic segments with endothelin-1, angiotensin II, serotonin and K+ after 16 h of ex vivo DOX treatment. DOX decreased VSMC contraction in response to 0.25 μM endothelin-1 (A) but not after stimulation with 2 μM angiotensin II (B), 2 μM serotonin (C) and 50 mM K+ (D). For (A–D), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. *, *** p < 0.05, 0.001; p > 0.05 in (B–D); For (A–C), n = 6 in each group; for (D), n = 10 in each group.
5. A Single Dose of DOX Decreases Ex Vivo Arterial Stiffness but Not In Vivo Arterial Stiffness and Blood Pressure
The possible translation of the aforementioned findings to arterial stiffness was investigated by evaluation of ex vivo and in vivo arterial stiffness.
The ROTSAC set-up was used for ex vivo evaluation of arterial stiffness, 16 h after in vivo DOX administration (4 mg/kg). Under Krebs Ringer conditions, the Peterson’s modulus (Ep), a measure for arterial stiffness, was lower in the DOX group, especially at higher pressures (Figure 6A). This effect was less pronounced in the presence of PE combined with L-NAME (Figure 6B). In the presence of DEANO, which removes VSMC tonus, DOX decreased Ep (Figure 6C) in a pressure-dependent way.
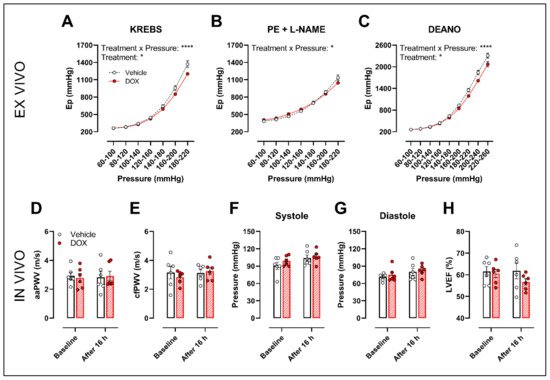
Figure 6. Evaluation of in and ex vivo aortic stiffness, 16 h after in vivo DOX administration. (A–C) Ex vivo evaluation of arterial stiffness with ROTSAC set-up. (D,E) In vivo evaluation of arterial stiffness with ultrasound imaging (D) and tonometry (E). (F) Systolic blood pressure measurements. (G) Diastolic blood pressure measurements. (H) Evaluation of LVEF with ultrasound imaging. Ep was lower in the DOX-treated group in a pressure-dependent way under Krebs Ringer conditions (A), but the effect was less pronounced in the presence of PE with L-NAME (B). Ep in response to DEANO was lower after DOX treatment (C). aaPWV (D), cfPWV (E), systolic blood pressure (F), diastolic blood pressure (G) and LVEF (H) were not affected after DOX treatment. For (A–H), repeated measures two-way ANOVA with Šidàk’s multiple comparisons test. *, **** p < 0.05, 0.0001; n = 6 for each group.
In vivo arterial stiffness was investigated by measuring the abdominal aorta pulse wave velocity (aaPWV) with ultrasound imaging as well as carotid–femoral pulse wave velocity (cfPWV) with tonometry, 16 h after injecting a single dose of DOX. Blood pressure and left ventricular ejection fraction (LVEF) were measured as well. DOX did not change aaPWV (Figure 6D), cfPWV (Figure 6E), blood pressure (Figure 6F,G) and LVEF (Figure 6H).
References
- Nagy, L.; Szabo, F.; Ivanyi, J.; Nemeth, L.; Kovacs, G.L.; Palatka, J.; Tarjan, J.; Toth, K.; Roth, E. A method for detection of doxorubicin-induced cardiotoxicity: Flow-mediated vasodilation of the brachial artery. Exp. Clin. Cardiol. 2001, 6, 87–92.
- Bosman, M.; Favere, K.; Neutel, C.H.G.; Jacobs, G.; De Meyer, G.R.Y.; Martinet, W.; Van Craenenbroeck, E.M.; Guns, P.D.F. Doxorubicin induces arterial stiffness: A comprehensive in vivo and ex vivo evaluation of vascular toxicity in mice. Toxicol. Lett. 2021, 346, 23–33.
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front. Pharmacol. 2019, 10, 1531.
- Duquaine, D.; Hirsch, G.A.; Chakrabarti, A.; Han, Z.; Kehrer, C.; Brook, R.; Joseph, J.; Schott, A.; Kalyanaraman, B.; Vasquez-Vivar, J.; et al. Rapid-onset endothelial dysfunction with adriamycin: Evidence for a dysfunctional nitric oxide synthase. Vasc. Med. 2003, 8, 101–107.
- Kalivendi, S.V.; Kotamraju, S.; Zhao, H.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J. Biol. Chem. 2001, 276, 47266–47276.
- Gibson, N.M.; Greufe, S.E.; Hydock, D.S.; Hayward, R. Doxorubicin-induced vascular dysfunction and its attenuation by exercise preconditioning. J. Cardiovasc. Pharmacol. 2013, 62, 355–360.
- Olukman, M.; Can, C.; Erol, A.; Oktem, G.; Oral, O.; Cinar, M.G. Reversal of doxorubicin-induced vascular dysfunction by resveratrol in rat thoracic aorta: Is there a possible role of nitric oxide synthase inhibition? Anadolu Kardiyol. Derg. 2009, 9, 260–266.
- Shen, B.; Ye, C.L.; Ye, K.H.; Zhuang, L.; Jiang, J.H. Doxorubicin-induced vasomotion and (i) elevation in vascular smooth muscle cells from C57BL/6 mice. Acta Pharmacol. Sin. 2009, 30, 1488–1495.
- Fransen, P.; Van Hove, C.E.; Leloup, A.J.; Martinet, W.; De Meyer, G.R.; Lemmens, K.; Bult, H.; Schrijvers, D.M. Dissecting out the complex Ca2+-mediated phenylephrine-induced contractions of mouse aortic segments. PLoS ONE 2015, 10, e0121634.
- van Langen, J.; Fransen, P.; Van Hove, C.E.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R.; Bult, H. Selective loss of basal but not receptor-stimulated relaxation by endothelial nitric oxide synthase after isolation of the mouse aorta. Eur. J. Pharmacol. 2012, 696, 111–119.
More