Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yury V. Zhernov and Version 2 by Lindsay Dong.
Eosinophilic esophagitis is a recently recognized allergic-mediated disease with eosinophil-predominant esophagus inflammation. Its pathogenesis is a complicated network of interactions and signaling between epithelial, mesenchymal, and immune cells on molecular and intercellular levels.
- food hypersensitivity
- eosinophilic esophagitis
1. Food Hypersensitivity Types Based on Molecular Mechanisms
FH is a broad term for an abnormal response related to food ingestion. Based on the pathophysiological mechanism of the reaction, food hypersensitivity can be divided into two broad categories [1][4]. The first category is immune-mediated reactions (i.e., FA). FA reactions are pathological immunologic responses to particular food antigens (called allergens) in a susceptible host. These reactions are reproducible each time the allergenic molecules (typically food protein antigens) are ingested. Based on the immunological mechanisms involved, FAs may be further classified into three types (Figure 1) [2][3].
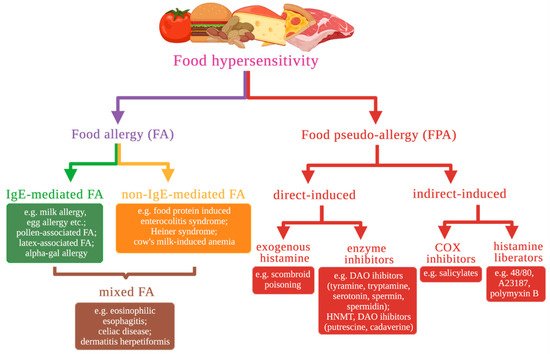
Figure 1. Classification of food hypersensitivity. Hypersensitivity reactions to food can be classified as food allergic (FA) or food pseudo-allergic (FPA) reactions. FA reactions are categorized further as IgE-mediated FA, non–IgE-mediated FA, or mixed FA (incl. eosinophilic esophagitis). FPA reactions are categorized further as direct-induced FPA or indirect-induced FPA. COX—cyclooxygenase, DAO—diamine oxidase, HNMT—histamine-N-methyltransferase.
IgE-mediated FA triggered when certain food allergen binds with allergen-specific antibodies that belong to the immunoglobulin E (IgE) class. It is the most common, best known, and well-characterized FA type. Typical IgE-mediated food allergic reactions occur immediately after allergen exposure, reproduce each time allergen is ingested, and are caused by food-specific IgEs, which can be detected using different approaches in order to diagnose FA and detect sensitization to specific allergen, i.e., persistence of allergen-specific IgEs [3][5]. This group includes milk allergy, egg allergy, pollen-associated FA, latex-associated FA, alpha-gal allergy, etc.
In sensitized individuals, allergen-specific IgE binds to FcƐRI with its Fc region. FcƐRI is the high-affinity IgE receptor. Mast cells, basophils, activated eosinophils, and some subtypes of antigen-presenting cells express FcƐRIα subunit on the surface of cellular membrane, allowing them to bind IgE molecules. To initiate downstream cellular signaling, multivalent allergen needs to bind with several IgEs-FcƐRIα complexes. Allergen initiates aggregation of IgEs-FcƐRIα recruits Lyn kinase, that phosphorylates FcεRIβ and γ subunits, allowing spleen tyrosine kinase (Syk) activation. Syk phosphorylates adaptor proteins, including linker for activation of T-cells (LAT). LAT recruits phospholipase PLCγ1, which produces second messengers: inositol 1,4,5-trisphosphate (IP3) and 2,3-diacylglycerol (DAG). IP3 activates exocytosis and degranulation of mast cells and basophils via increase of intracellular Ca2+ level. DAG activates protein kinase C, which phosphorylates myosin light chain to transport granules to cellular membrane. Mediators in mast cell and basophil granules (histamine, serotonin, serine proteases and others), when released, cause vasodilatation, smooth muscle constriction, increase vascular permeability, producing FA symptoms, e.g., urticaria, angioedema, diarrhea and vomiting, bronchospasm, hypotension [4][6].
In non-IgE FA, specific IgE to food antigens are not involved in an allergic reaction. Cellular mechanisms of the immune response and type II and III hypersensitivity are responsible for FA. Non-IgE FA includes various disorders such as food protein-induced enterocolitis syndrome (FPIES), Food Protein-Induced Allergic Proctocolitis (FPIAP), Heiner’s syndrome, cow’s milk-induced anemia, etc.
In mixed FA, both antigen-specific IgE and immune cells are involved in the reaction. Mixed FA has been increasing worldwide [5][7]. The most common mixed FAs are Eosinophilic Esophagitis (EoE) and Non-EoE eosinophilic gastrointestinal disorders (Non-EoE-EGID), which may occur as eosinophilic gastritis, colitis, or gastroenteritis.
The second category is FPA, which is similar to true allergies but differ from FA in that they are not a consequence of a dysregulation of the immune system. FPA occurs due to the properties of the food itself and the abnormal response of the host. The first may be due to components in food products that may be either exogenous or present naturally in food. Abnormal responses of the host include functional nontoxic and nonimmunologically mediated reactions. Natural and artificial organic compounds may cause adverse food reactions in sensitive people if consumed sufficiently; the degree of sensitivity varies between individuals.
FPA can be divided into two groups depending on the induction of histamine release. Direct-induced FPA is manifested by the action of exogenous histamine (from histamine-rich foods) [6][8] or enzyme inhibitors (tyramine, tryptamine, putrescine, etc.) that initiate the release of histamine from cells. Metabolism disorders are most commonly attributed to diamine oxidase (DAO) enzyme deficiency. Less common causes are histamine-N-methyltransferase (HNMT) and aldehyde oxidase (AOX1) deficiencies, which alter intracellular histamine breakdown.
Indirect-induced FPA is caused by foods containing histamine-releasing agents–COX inhibitors or histamine liberators. Salicylate–containing foods cause FPA reactions [7][9] based on the inhibition of cyclooxygenase–1 (COX1) by salicylates from natural food sources. COX1 inhibition results in reduced arachidonic acid use in the prostaglandin synthesis pathway. In intolerant individuals, this leads to activation of the leukotriene metabolism with increased formation of LTB4 and/or LTC4–E4. Typical symptoms of salicylate intolerance are respiratory complaints, including asthma and sinus inflammation with recurring nasal polyps, known as Samter’s Triad [8][10]. Sometimes, symptoms may include gastrointestinal complaints with meteorism, flatulence, diarrhea, and, rarely, colitis with strictures and ulcers [9][11]. Histamine liberators can induct release histamine from mast cells and eosinophils without binding to cell receptors. Compound 48/80, ionophore A23187 can cause significant histamine release. In each case, a release is triggered by an increase in levels of free cytosolic calcium [10][12]. However, the pathogenesis of the action of histamine liberators is even less clear.
 In situ hybridization on esophageal biopsy specimens identified the esophageal epithelium as the main source of Eotaxin–3 production [13][32]. Primary esophageal epithelial cells stimulated with IL–13 produced transcriptional changes largely overlapped with the EoE transcriptome (22% of IL–13–induced genes were present in the EoE transcriptome) [14][33]. CCL26 was the most upregulated gene in the IL–13–stimulated esophageal cells [14][33]. IL–13 and IL–4 activate signal transducer and activator of transcription 6 (STAT6) [20][39]. The STAT6 binding site (–55 to –64) is located upstream of the CCL26 transcription initiation site and is required for IL–13–induced CCL26 promoter activity in esophageal epithelial cells [14][23][33,42]. In addition, the cAMP-response element (CRE) site in the CCL26 promoter (–230 to –237) acts in concert with the STAT6 site [23][42] and functions as a transcriptional coactivator for STAT6 [24][43]. ChIP analysis has shown that STAT6 binds to the CCL26 promoter and recruits CRE-binding protein (CREB) binding protein (CBP) following stimulation with IL–13 [23][42]. CBP activates basal and IL–13–induced CCL26 promoter activity. CBP acetylates histone protein H3 at the transcription start site (TSS) and promotes an open chromatin structure facilitating CCL26 transcription [23][42]. Moreover, higher levels of H3 acetylation were observed in the esophageal tissue in EoE compared to the control, which may be partly attributed to the IL–13–dependent activation of CBP intrinsic histone acetyltransferase activity [23][42].
In situ hybridization on esophageal biopsy specimens identified the esophageal epithelium as the main source of Eotaxin–3 production [13][32]. Primary esophageal epithelial cells stimulated with IL–13 produced transcriptional changes largely overlapped with the EoE transcriptome (22% of IL–13–induced genes were present in the EoE transcriptome) [14][33]. CCL26 was the most upregulated gene in the IL–13–stimulated esophageal cells [14][33]. IL–13 and IL–4 activate signal transducer and activator of transcription 6 (STAT6) [20][39]. The STAT6 binding site (–55 to –64) is located upstream of the CCL26 transcription initiation site and is required for IL–13–induced CCL26 promoter activity in esophageal epithelial cells [14][23][33,42]. In addition, the cAMP-response element (CRE) site in the CCL26 promoter (–230 to –237) acts in concert with the STAT6 site [23][42] and functions as a transcriptional coactivator for STAT6 [24][43]. ChIP analysis has shown that STAT6 binds to the CCL26 promoter and recruits CRE-binding protein (CREB) binding protein (CBP) following stimulation with IL–13 [23][42]. CBP activates basal and IL–13–induced CCL26 promoter activity. CBP acetylates histone protein H3 at the transcription start site (TSS) and promotes an open chromatin structure facilitating CCL26 transcription [23][42]. Moreover, higher levels of H3 acetylation were observed in the esophageal tissue in EoE compared to the control, which may be partly attributed to the IL–13–dependent activation of CBP intrinsic histone acetyltransferase activity [23][42].

2. Eosinophilic Esophagitis
Eosinophilic esophagitis (EoE) is a recently recognized allergic-mediated disease with eosinophil-predominant esophagus inflammation. Clinically, it is characterized by various symptoms related to esophageal dysfunction, including vomiting, regurgitation, feeding difficulties, heartburn, failure to thrive in infants, dysphagia, or food bolus impaction [11][13]. Symptoms are nonspecific and mimic those observed in gastroesophageal reflux disease (GERD).2.1. The Role of the Eotaxin-3 and IL-13 in the Development of EoE
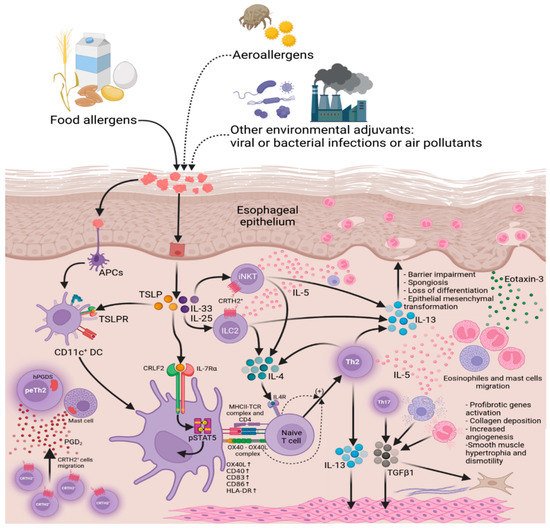
The CCL26 gene has the largest fold change in mRNA expression level between EoE transcriptome and controls in many studies [12][13][14][31,32,33]. The CCL26 codes chemokine Eotaxin-3, implicated in eosinophil trafficking to the esophagus in patients with EoE via chemokine receptor CCR3. Of the eotaxins, CCL26/Eotaxin-3 is the most upregulated in patients with EoE, and its expression correlates with eosinophil (and mast cell) levels within esophageal biopsy specimens, indicating a specific contribution in the disease. Only modest changes of other eotaxin family genes (CCL11/Eotaxin–1 and CCL24/Eotaxin-2) were observed in EoE patients. The mouse homolog of CCL26 is a pseudogene [15][34], although CCR3–deficient mice were nearly wholly protected from the development of esophageal eosinophilia in the experimental EoE model [13][32]. Levels of CCL26 transcript in a single biopsy specimen are susceptible in distinguishing EoE from control populations [16][35] and GERD patients [17][36], despite the histological “patchiness” of EoE across multiple biopsy specimens. Studies have determined that TH2–derived interleukin IL–13 is one of the critical signaling molecules altering gene expression in EoE. It is well established that IL–13 is overproduced in EoE patients’ biopsy specimens. IL–13 mRNA expression in active EoE by RT–PCR was 16–fold higher compared to healthy controls [14][16][33,35]. In contrast, the IL–4 mRNA level was not significantly increased in EoE. Still, a statistically significant difference in IL–4 expression was observed between EoE patients with and without atopic comorbidities, with higher IL–4 expression levels in atopic individuals [16][35]. The CC10–rtTA–IL13–transgenic mouse is a well–characterized model of asthma [18][37]. These mice contain transgenic construct that makes possible external regulation of IL13 gene expression in lung tissue. IL–13 overexpression with the CC10–rtTA–IL13 transgenic system in response to exogenous doxycycline is sufficient to induce alteration resembling EoE, i.e., esophageal eosinophilia, tissue remodeling of the esophagus: increased esophageal circumference, increased epithelial cell proliferation primarily associated with the basal zone, collagen deposition, and increased angiogenesis in the lamina propria [19][38]. IL–13–induced changes in murine esophageal transcriptome significantly overlap with human EoE transcriptome data [19][38], including murine Eotaxin–1 and Eotaxin–2 esophageal production. Esophageal epithelial cells express all components of the IL–13 receptor, including IL–4Rα, IL–13Rα1, and IL–13Rα2 [20][39]. IL–13 is produced by Th2 cells [21][40], activated eosinophils [22][41], ILC2, and iNKT cells (Figure 2).Figure 2. Cellular mechanisms of eosinophilic esophagitis pathogenesis. Allergens/adjuvants (incl. food allergens) stimulate the esophageal epithelium by inducing thymic stromal lymphopoietin (TSLP) and Interleukin (IL)–33, leading to stimulation of T helper 2 cells (Th2), natural killer cells (NK cells), mast cells, basophils, and type 2 innate lymphoid cells (ILC2). NK cells, mast cells, basophils, ILC2, and Th2 cells induce IL–4, which induces Th2 differentiation. IL-4 and IL-13 induced by Th2 cells provoke the release of Eotaxin–3, which stimulates eosinophils to secrete IL–5. IL–5 secreted by Th2 cells and mast cells also stimulate eosinophils. Mast cells, eosinophils, Th2 cells induce transforming growth factor beta 1 (TGFβ1), stimulating eosinophils and fibroblasts. Th2 cells also induce IL–13, which causes impaired barrier function and tissue alteration. APCs—antigen-presenting cells, TSLPR—thymic stromal lymphopoietin receptor, DC—dendritic cells, peTh2—pathogenic effector Th2 cells, CRTH2—prostaglandin D2 receptor 2, hPGDS—human prostaglandin D synthase, CRLF2—cytokine receptor-like factor 2, pSTAT5—phosphorylated signal transducer and activator of transcription 5, MHCII—major histocompatibility complex class II, TCR—T-cell receptor, OX40L—ligand for OX40, iNKT—invariant natural killer T-cells.
2.2. Impairment of Esophageal Epithelium Barrier Function
3.2. Impairment of Esophageal Epithelium Barrier Function
The prominent pathophysiological feature of EoE is impairment of esophageal epithelium barrier function (BF). The healthy esophageal epithelium protects against adverse environmental factors, including food antigens and gastric acid refluxate, that are able to penetrate into esophageal tissue causing structural damage or inflammatory responses. The vast body of evidence suggests that barrier dysfunction and allergic inflammation-related changes in the esophageal epithelium are essential processes in EoE pathology. The morphology of the inflamed esophageal epithelium in EoE has several features, including basal zone hyperplasia in the esophageal epithelium, which replaces much of the more differentiated upper layer of epithelial cells, and the emergence of DIS in the suprabasal layers, which is believed to be associated with an increase in permeability of esophageal epithelium to food allergens, refluxed acid, microbes, and other alternating factors in EoE. On the molecular level, epithelial BF depends on the proper expression of structural genes coding proteins that comprise multiprotein complexes called cell junctions. Cell junctions provide adhesion between two cells (e.g., between neighboring epitheliocytes in esophageal epithelium) or cell and extracellular matrix proteins. Cell junctions are involved in intracellular signaling. Cell junction proteins’ dysregulation leads to loss of adhesion between cells impairing epithelial BF and increasing paracellular permeability, and at the same time dysregulates signal transduction in the cells. Proteins of tight junctions (claudins 1 and 7 and occludin) [25][26][27][45,46,47], adherens junctions (E–cadherin), and desmosomes (DSG1) [28][25][44,45] are shown to be downregulated in EoE.2.3. The Role of the Cadherin 26 in the Development of EoE
3.3. The Role of the Cadherin 26 in the Development of EoE
However, the recently characterized cadherin 26 (CDH26) is highly upregulated in both active EoE [14][33] and eosinophilic gastritis (EG) [29][30][48,49] patients. It was the only intersecting upregulated gene in these data. CDH26 gene expression is upregulated in epithelial cells by Th2 cytokines [30][49]. Immunohistochemical staining of control group biopsies with CDH26-specific antibodies revealed that CDH26 expressed in superficial layers of esophageal epithelium. Staining in active EoE specimens covered both epithelial cells of superficial layers and basal zone cells. Intensity of CDH26 staining in active EoE biopsies surpassed control levels of CDH26 [30][49]. By Western blot analysis, CDH26 had a 4.9–fold increase in EG and 3.4–fold increase in EoE compared to the control, so CDH26 is highly upregulated in esophageal and gastric tissues under allergic inflammation [30][49]. CDH26 exhibits sequence homology to the cadherin family of proteins, with five extracellular cadherin repeats [31][50]. CDH26 has been shown to localize on the cell surface membrane of esophageal epithelial cells and be modified by N–linked glycosylation of asparagine residues [30][49]. Co–immunoprecipitation shows that CDH26 is a functional cadherin that interacts in a homotypic manner with other CDH26 molecules, mediates calcium–dependent cell adhesion, dimerizes or multimerizes, and interacts with α–, β–, and p120–catenins [30][32][49,51]. CDH26 also binds α4 and αE integrins that are co-immunoprecipitated with CDH26 [30][49]. The recombinant CDH26–hIgG1–Fc antibody binds α4β7 integrin, CDH26–expressing cells adhere to integrin α4β7–coated surface, and Jurkat cells that express integrin α4β1 [33][52] adhere to recombinant CDH26–hIgG1–Fc in an integrin α4–dependent manner, so, CDH26 is proposed to have the ability to impact diverse α4+ and/or αE+ cells (e.g., CD4+ T cells, eosinophils, and mast cells) migration and adhesion [30][49]. Altered intraepithelial localization of several subsets of cells in EoE correlates with this fact.2.4. The Role of the Desmosomal Cadherin Desmoglein-1 in the Development of EoE
3.4. The Role of the Desmosomal Cadherin Desmoglein-1 in the Development of EoE
The desmosomal cadherin desmoglein–1 (DSG1) is one of cell adhesion molecules, glycoprotein assigned to the cadherin superfamily. DSG1 is an essential component of desmosomes, and forms cell-to-cell junctions in epithelia, e.g., in epidermis. DSG1 is considered to be involved in pathogenesis of atopic diseases, e.g., homozygous mutations in DSG1 causes severe dermatitis, multiple allergies, and metabolic wasting (SAM syndrome); autoimmunization against DSG1 causes pemphigus foliaceus, a skin blistering disease, which manifests as severe loss of epithelial integrity, compromised BF and skin lessions [28][44]. There is a substantial decrease (12.7–fold reduction in RNA–seq and 22.1–fold reduction in RT–PCR) in the expression of DSG1 in the esophageal biopsies of patients with active EoE [28][44]. The downregulation of DSG1 was specific among other DSG family members, including DSG3, the most abundant in esophageal mucosa DSG. Immunofluorescent and immunohistochemical staining for DSG1 revealed that expression of this protein is mainly localised to suprabasal layers of esophageal epithelium in control biopsy samples. In active EoE, pronounced loss of DSG1 expression was observed [28][44]. DSG3 and E–cadherin were unchanged between the control and active EoE in this study [28][44]. The reduction in DSG1 levels is consistent with a significant decrease in the number of desmosomes per cell, which is a distinctive ultrastructural feature of active EoE compared with inactive EoE, GERD, and normal epithelia [34][61]. Facts suggest that impairment of BF in EoE may be caused by other alterations in cellular contacts, e.g., observed in the EoE loss of DSG1. Experiments with DSG1 gene shRNA silencing in ALI–differentiated EPC2 cells reproduced this impairment in BF. shRNA–transduced cultures demonstrated impaired TER (42% decrease) and increased FITC–dextran paracellular flux (33%) [28][44]. So, DSG1 loss is sufficient to impair esophageal epithelium BF in vitro, and DSG1 mRNA and protein decrease in patient biopsies has a significant contribution to BF impairment in EoE [28][44].2.5. Loss of Esophageal Epithelium Differentiation
3.5. Loss of Esophageal Epithelium Differentiation
IL–13 contribution to EoE pathogenesis beyond Eotaxin–3 overproduction includes profound dysregulation of the epidermal differentiation complex (EDC) gene expression [35][62]. The epidermal differentiation complex (EDC) is a gene complex on the human chromosome 1 in a locus 1q21. Genes residing in EDC have similar, closely related functions and are essential for epithelial barrier formation and expressed during maturation of epithelial cells terminal differentiation [36][63]. Across the human genome, the highest density of genes, expression of which is dysregulated in active EoE, is observed to occur in EDC locus [35][62]. Expression patterns in esophageal biopsy specimens of EoE patients show significant decreased expression or trends toward the decreased expression of most genes in the EDC locus [35][62]. Ex vivo response to IL–13 presents a similar downregulation of EDC genes, including filaggrin (FLG), involucrin (IVL), and several small proline–rich repeat (SPRR) family members (1A, 2D, 3, and 4) [35][62]. FLG is expressed in the skin epidermis and epithelium of esophageal, nasal and oral mucous membranes. FLG encodes progenitor protein Profilaggrin. During epithelial cell differentiation, Profilaggrin undergoes processing, and after proteolytic cleavage Filaggrin monomers are formed. Filaggrin is one of the essential structural proteins for stratified epithelial BFs. Filaggrin function is aggregation of keratin intermediate filaments during transformation of granular cells into flattened squamous cells, that compose the superficial layer of esophageal epithelium and essential for its BF, despite the fact that actual keratinization normally does not occur in esophagus, and human esophageal epithelium is stratified, squamous and nonkeratinized [37][64]. In the case of FLG downregulation, epithelial BF decreases, and exogenous allergens become able to penetrate epithelial barriers and activate immune responses. Loss of FLG and other EDC gene expression leads to defects in epidermal BF. FLG and IVL expression in EoE biopsies is decreased on gene and protein level [38][65]. IL-13 also decreases levels of FLG and IVL mRNAs and proteins in ALI-cultured primary human esophageal epithelial cells (HEEC) [38][65]. Furthermore, FLG silencing with siRNA in ALI HEEC causes BF impairment; TEER and thickness of the cell layer was decreased in siRNA-transfected cultures, indicating alterations in cell proliferation and differentiation [38][65]. It is remarkable that tight junction proteins (CLDN1 and CLDN4) have altered patterns of expression in FLG-deficient cells, although the levels of the proteins are unchanged [38][65].2.6. The Role of the CAPN14 in the Development of EoE
3.6. The Role of the CAPN14 in the Development of EoE
CAPN14 is a cytosolic calcium–activated cysteine protease that was identified as an associated locus 2p23 in EoE genome-wide association studies (GWAS) [39][40][71,72]. In comparison with other members of the calpain family, CAPN14 possesses a unique feature of its tissue-specific expression pattern. CAPN14 is almost specifically expressed in the esophageal epithelium [40][72]. Stimulation of EPC2 esophageal epithelial cells with IL-13 significantly upregulates CAPN14 expression. IL-13 impact on calpain family gene expression is confined to CAPN14 upregulation; other calpains except CAPN14 are not induced by IL-13 in primary esophageal epithelial cell culture and in EPC2 ALI cultures [40][41][72,73]. However, in specimens of esophageal biopsies, in addition to CAPN14 upregulation, CAPN3 level turned out to be significantly elevated. Conversely, expression of CAPN7, CAPN5, CAPNS2 and CAST (calpastatin, endogenous calpain inhibitor) genes were downregulated [40][72]. Throughout the calpain family and related genes, CAPN14 reveals highest fold change. The kinetics of IL-13-induced CAPN14 expression are parallel to the induction of CCL26 in EPC2 cells [42][74]. Pronounced changes in an epigenetic signature are observed in the promoter region of CAPN14 in response to IL–13 stimulation. The ChIP–seq detected a marked increase in H3 acetylation at the 27th lysine residue (H3K27Ac) [40][43][72,75] and H3 trimethylation at the 4th lysine residue (H3K4me3) [43][75] in the CAPN14 promoter region near the TSS in IL–13–treated cells. H3K27Ac and H3K4me3 are highly enriched at active promoters near TSS and positively correlated to gene transcription, which is consistent with an increase in CAPN14 transcriptional activity by RNA–seq [40][72].2.7. The Role of the POSTN in the Development of EoE
3.7. The Role of the POSTN in the Development of EoE
Moreover, DSG1 deficiency increases gene expression of the proinflammatory extracellular matrix molecule periostin (POSTN) [44][76]. POSTN is one of the markedly upregulated genes in EoE transcriptome (35-fold change), that encodes periostin—protein of extracellular matrix (ECM), that facilitates epithelial-mesenchymal transition (EMT), fibrotic remodeling and migration of certain cells to inflamed tissues. Periostin can directly enhance activated eosinophil adhesion via integrin αMβ2 [45][77], as well as increase keratinocyte production of thymic stromal lymphopoietin (TSLP), a potent Th2–skewing cytokine [46][78] that has been associated with EoE [47][48][79,80]. It has been shown that treatment of esophageal epithelial cells with IL–13 induces POSTN expression in this cell type [49][81], as well as in bronchial epithelial cells [50][82]. POSTN can induce epithelial–mesenchymal transition by increasing signaling through integrin αVβ5 and epidermal growth factor receptor (EGFR) [51][83]. It is possible because epitheliocytes adopt a fibroblast-like phenotype due to induced loss of epithelial cell markers [52][84]. Moreover, it has been demonstrated that DSG1–dependent EGFR signaling suppression promotes epithelial differentiation and reduces proliferative capacity [53][85]. The epithelial–mesenchymal transition has been proposed to occur in EoE (Figure 3) [54][55][86,87].Figure 3. The signaling pathways of the interleukin–13 receptor (IL13R), transforming growth factor beta (TGFβ) receptor, and epidermal growth factor receptor (ErbB) in esophagus keratinocyte and their alterations in eosinophilic esophagitis. The IL–13R receptor binds to its corresponding ligand, and heterodimerization occurs, enhancing Janus kinase (JAK) activity. Signaling molecules such as signal transducer and transcriptional activator (STAT) 6 and STAT3 can initiate transcription of target genes, including eotaxin–3. The effects of IL–13 are mediated by ErbB. ERBB2–Erbb2 interacting protein (ERBIN) negatively regulates TGFβ signaling. TGFβ mediates fibrosis by inducing fibrogenic target genes. Active TGFβ binds to its receptor to initiate SMAD–dependent and independent signaling. SMAD–dependent signaling regulates fibrogenic target genes such as α–smooth muscle actin, collagen, connective tissue growth factor, tissue metalloprotease inhibitor, and periostin. TYK2—non-receptor tyrosine-protein kinase, CRE—cAMP response element, CREB—cAMP response element-binding protein, CBP—CREB-binding protein, CoA—Coenzyme A, Ac—acyl group, H3K27ac—lysine acetylation at N-terminal position 27 of histone H3, CCL26—chemokine (C-C motif) ligand 26, CAPN14—calpain-14, POSTN—periostin, DSG1—desmoglein 1, SHOC2—Leucine-rich repeat protein SHOC-2, ErbB—receptor tyrosine-protein kinase ErbB, TGFβ—transforming growth factor β, MEK—mitogen-activated protein kinase kinase, ERK—extracellular signal-regulated kinase.
3.8. EoE-Associated Risk Genes
2.8. EoE-Associated Risk Genes
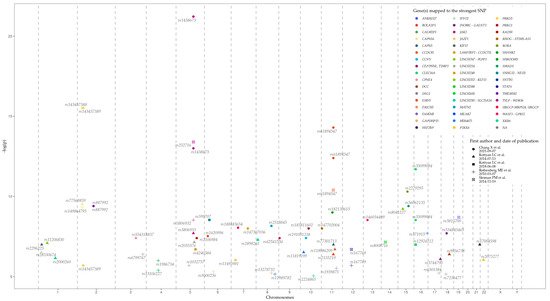
Loci, genes, and SNPs that have the most significant association with EoE on the basis of GWAS data are represented in Table 1 and Figure 4. Data were retrieved from the GWAS Catalog (EFO ID: EFO_0004232). The biological meaning and role in EoE predisposition and development of many genes, represented in the table, remain obscure. Further studies are needed to identify the influence of associated variations, genes, and their products, on EoE pathogenesis.
Figure 4. The position of the single nucleotide polymorphisms (SNP) with the most significant association with EoE from the genome-wide association studies (GWAS) data on the human genome.
Table 1. Genome–wide significant loci reported in GWAS.
| EoE Risk Locus | Mapped Gene | Tag SNP | The Strongest SNP Risk Allele | p-Value | OR | Reference |
|---|---|---|---|---|---|---|
| 1p13.3 | LINC02785 SLC25A24 |
rs2000260 | A | 7 × 10−7 | 1.32 | [40] |
| 1p32.2 | LINC01767 PLPP3 |
rs11206830 | ? | 8 × 10−8 | 2.162 | [40] |
| 1p36.12 | KIF17 | rs2296225 | ? | 1 × 10−7 | 1.626 | [40] |
| 1p36.13 | IFFO2 | rs28530674 | ? | 3 × 10−7 | 1.826 | [40] |
| 2p22.2 | PRKD3 | rs143457389 | A | 3 × 10−16 | 1.77 | [56] |
| 2 × 10−6 | 1.91 | |||||
| 2p23.1 | CAPN14 | rs143457388 | A | 3 × 10−16 | 1.77 | [56] |
| rs149864795 | A | 5 × 10−10 | 2.216 | [39] | ||
| rs77569859 | G | 3 × 10−10 | 1.98 | [40] | ||
| 2q12.1 | TMEM182 | rs887992 | C | 4 × 10−10 | 0.75 | [56] |
| 3q22.1 | CPNE4 | rs554318837 | C | 4 × 10−8 | 2.88 | [56] |
| 3q26.32 | ? | rs6799767 | ? | 4 × 10−7 | 1.49 | [57] |
| 4q21.1 | SHROOM3 | rs13106227 | ? | 4 × 10−6 | 1.52 | [47] |
| rs1986734 | ? | 1 × 10−6 | 1.54 | |||
| 5q14.2 | ? | rs1032757 | T | 2 × 10−6 | 1.96 | [47] |
| 5q22.1 | TSLP | rs3806932 | ? | 3 × 10−9 | 1.85 | [47] |
| rs3806933 | G | 2 × 10−8 | 1.37 | [40] | ||
| TSLP WDR36 |
rs252716 | C | 4 × 10−14 | 1.516 | [39] | |
| WDR36 RPS3AP21 |
rs1438673 | C | 1 × 10−13 | 1.43 | [56] | |
| 6 × 10−22 | 0.7 | |||||
| 5q23.1 | LINC02214 | rs2055376 | A | 7 × 10−8 | 2.3 | [40] |
| 5q23.2 | LINC02240 | rs4240384 | ? | 2 × 10−7 | 1.4326648 | [57] |
| 5q31.1 | RAD50 | rs2106984 | A | 4 × 10−8 | 1.26 | [56] |
| 6p11.2 | GAPDHP15 | rs9500256 | ? | 5 × 10−6 | 2.04 | [47] |
| 6p21.33 | SNHG32 NEU1 |
rs599707 | ? | 3 × 10−9 | 1.6920472 | [57] |
| 6p22.3 | BOLA2P3 | rs1620996 | T | 3 × 10−8 | 0.69 | [56] |
| 7p13 | URGCP-MRPS24 URGCP |
rs188483654 | C | 9 × 10−9 | 5.68 | [56] |
| 7p15.1 | JAZF1 | rs11495981 | ? | 9 × 10−7 | 1.308 | [57] |
| 7q22.3 | LARP1BP2 CCDC71L |
rs147307036 | A | 1 × 10−8 | 8.04 | [56] |
| 8p23.1 | XKR6 | rs2898261 | C | 5 × 10−8 | 1.35 | [40] |
| 8q22.2 | MATN2 | rs2513845 | T | 7 × 10−9 | 4.18 | [56] |
| ERICH5 | rs13278732 | T | 6 × 10−6 | 1.31 | [47] | |
| 8q24.12 | SNTB1 | rs11989782 | A | 7 × 10−6 | 1.53 | [47] |
| 9p24.1 | JAK2 | rs62541556 | T | 4 × 10−8 | 1.61 | [56] |
| 10p11.21 | CCNY | rs191051238 | C | 4 × 10−8 | 13.2 | [56] |
| 10p12.31 | MIR4675 | rs11819199 | G | 3 × 10−7 | 1.62 | [40] |
| 10q21.1 | PRKG1 | rs185811602 | T | 1 × 10−8 | 6.37 | [56] |
| 10q23.1 | LINC02650 | rs2224865 | G | 9 × 10−6 | 1.44 | [47] |
| 11p15.4 | RHOG STIM1-AS1 |
rs147702004 | T | 1 × 10−8 | 1.95 | [56] |
| 11q13.4 | SHANK2 | rs182139615 | T | 1 × 10−9 | 6.62 | [56] |
| 11q13.5 | EMSY | rs61894547 | T | 4 × 10−11 | 2.439 | [39] |
| T | 4 × 10−13 | 1.92 | [56] | |||
| T | 5 × 10−15 | 1.79 | ||||
| EMSY LINC02757 |
rs2155219 | A | 4 × 10−7 | 1.37 | [40] | |
| CAPN5 | rs77301713 | ? | 1 × 10−7 | 2.22 | [40] | |
| 11q14.2 | CCDC81 | rs118086209 | C | 2 × 10−7 | 2.19 | [40] |
| 11q21 | FAM76B | rs1939875 | T | 3 × 10−6 | 1.54 | [47] |
| 12q13.3 | STAT6 | rs167769 | T | 2 × 10−7 | 1.351 | [39] |
| T | 2 × 10−6 | 1.36 | [47] | |||
| 13q12.13 | WASF3 GPR12 |
rs146034499 | A | 3 × 10−9 | 5.92 | [56] |
| 14q12 | LINC02588 | rs8008716 | G | 7 × 10−8 | 1.712 | [39] |
| 15q13.3 | LINC02352 KLF13 |
rs8041227 | G | 6 × 10−12 | 1.52 | [40] |
| 15q22.2 | RORA | rs2279293 | G | 5 × 10−11 | 0.69 | [56] |
| 15q22.33 | SMAD3 | rs56062135 | T | 4 × 10−12 | 1.29 | [56] |
| 16p13.13 | CLEC16A | rs35099084 | C | 3 × 10−9 | 0.71 | [56] |
| T | 2 × 10−12 | 0.72 | ||||
| rs12924112 | ? | 1 × 10−7 | 1.310616 | [57] | ||
| 16q24.1 | MEAK7 | rs371915 | ? | 2 × 10−8 | 1.9 | [47] |
| 17q24.3 | CALM2P1 | rs6501384 | T | 6 × 10−6 | 1.41 | [47] |
| 17q25.3 | CEP295NL TIMP2 |
rs3744790 | ? | 8 × 10−7 | 1.54 | [40] |
| 18q12.1 | DSG1 | rs7236477 | G | 7 × 10−6 | 2.22 | [47] |
| 18q12.2 | INO80C GALNT1 |
rs534845465 | A | 2 × 10−8 | 5.78 | [56] |
| DCC | rs9956738 | ? | 4 × 10−7 | 2.472 | [40] | |
| 19q13.11 | ANKRD27 | rs3815700 | C | 2 × 10−9 | 1.618 | [39] |
| 21q22.3 | HSF2BP | rs17004598 | C | 1 × 10−7 | 2.57 | [40] |
| 22q11.21 | P2RX6 | rs2075277 | ? | 9 × 10−7 | 1.544 | [40] |
34. Conclusions
EoE pathogenesis is a complicated network of interactions and signaling between epithelial, mesenchymal, and immune cells on molecular and intercellular levels. Alterations produced by overactivation of some cytokine signaling pathways, e.g., IL–13 or TSLP, were evolved and observed in this review from the viewpoints of molecular, genetic, epigenetic, and transcriptomic changes. Despite the substantial amount of experimental data, the reliable and representative mechanism of EoE pathogenesis has yet to show itself, and so the place of EoE between mixed and non-IgE-mediated allergic disorders, between eosinophilic gastrointestinal disorders currently seems vague and unclear.