MERTK and AXL are members of the TAM family of receptor tyrosine kinases and are abnormally expressed in 69% and 93% of non-small cell lung cancers (NSCLCs), respectively. Expression of MERTK and/or AXL provides a survival advantage for NSCLC cells and correlates with lymph node metastasis, drug resistance, and disease progression in patients with NSCLC. The TAM receptors on host tumor infiltrating cells also play important roles in the immunosuppressive tumor microenvironment. Thus, MERTK and AXL are attractive biologic targets for NSCLC treatment.
- MERTK
- AXL
- TAM family
- NSCLC
1. Introduction
Lung cancers are divided into two broad categories: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Following clinical validation of translational inhibitors targeting two important NSCLC oncogenic drivers, epidermal growth factor receptor (EGFR) [15,16,17,18][1][2][3][4] and anaplastic lymphoma kinase (ALK) [19,20,21][5][6][7], molecular-targeted therapies have been applied to the management of metastatic NSCLC, resulting in remarkably improved prognosis and quality of life relative to patients treated with conventional chemotherapeutics [22,23,24][8][9][10]. Additional mutated oncogenic proteins have been identified in NSCLC, including HER2, BRAF, RET, MET, and ROS1 [20,25][6][11]. Even though patients respond to targeted therapies initially, the majority of patients, if not ultimately all patients, relapse within 1 to 2 years when treated with targeted therapies [26,27,28,29,30][12][13][14][15][16]. Therefore, understanding the mechanisms of primary and secondary resistance to current targeted therapies is critical to enhance patient outcomes. New therapeutic approaches will be required to further enhance outcomes. Both MERTK and AXL, members of the TAM (TYRO3, AXL, and MERTK) family of receptor tyrosine kinases (RTK), are emerging therapeutic targets in NSCLC.
2. Physiologic Roles for MERTK and AXL
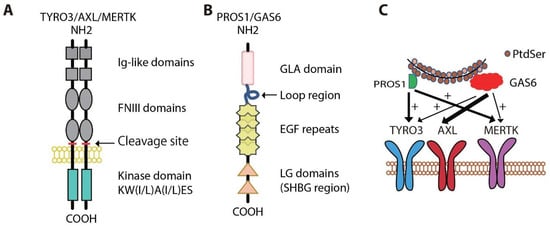
3. Oncogenic Roles for MERTK and AXL
3.1. Roles in NSCLC
MERTK and AXL are frequently aberrantly expressed in NSCLC patient samples, but are absent or expressed at low levels in normal human bronchial epithelial cells [51,52,53,54,55,56,57,58][37][38][39][40][41][42][43][44]. High levels of AXL have been described in subsets of both treatment-naïve and relapsed NSCLC [58,59,60][44][45][46]. Increased AXL expression was associated with increased tumor cell invasiveness and tumor grade and predicted poorer survival in patients with NSCLC [56,57,61,62,63,64,65][42][43][47][48][49][50][51]. Inhibition of MERTK in NSCLC cell lines with a small molecule MERTK tyrosine kinase inhibitor (TKI), MERTK-specific blocking monoclonal antibody, or shRNA induced apoptosis and decreased colony formation in vitro and inhibited tumor growth in vivo [66,67,68,69][52][53][54][55]. Treatment with an antibody against active AXL or siRNA/shRNA AXL knockdown also provided anti-tumor activity in NSCLC models [56,64,70][42][50][56].
High levels of MERTK and/or AXL have also been implicated in drug resistance and radioresistance [54,55,60,69,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86][40][41][46][55][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72]. Increased MERTK or AXL expression in NSCLC correlated with chemotherapy resistance [51,66,70,87,88,89][37][52][56][73][74][75]. Conversely, MERTK or AXL knockdown, treatment with a MERTK monoclonal antibody, or AXL inhibitor R428 or MP-470 promoted apoptosis and increased the sensitivity of NSCLC cells to chemotherapeutic agents [51,66,70,87,88,89][37][52][56][73][74][75]. Upregulated AXL and its interaction with EGFR were associated with resistance to PI3Kα inhibition due to sustained mTOR activation, and addition of AXL inhibitor R428 sensitized tumor cells to PI3Kα [76][62]. In addition, AXL has been implicated in resistance to anti-IGF-1R therapy [90,91][76][77] and resistance to BRAF/MEK inhibitors [92][78].
3.2. Functions in Cancer Cells
3.3. Signaling in Cancer Cells
Activation of AXL and/or MERTK leads to signaling cascades that are important for tumor progression (Figure 2). MERTK kinase activity is associated with phosphorylation at three tyrosine residues: Y749, Y753, and Y754 on MERTK [131][98] and Y779, Y821, and Y866 on AXL [132,133][99][100]. Both Y779 and Y821 on AXL and two additional phosphorylation sites for MERTK (Y872 and Y929) are docking sites for GRB2 and the p85 regulatory subunit of PI3K, which activate MEK/ERK and PI3K/AKT signaling pathways, respectively [109,132,133][82][99][100]. The MEK/ERK signaling is associated with cell proliferation [134][101], while the PI3K/AKT pathway is preferentially involved in tumor cell survival [135][102]. MERTK-dependent cell migration is mediated by FAK signaling [55[41][103],136], while MERTK induced transformation correlates with activation of STAT-dependent transcription [137][104]. The anti-apoptotic effects of MERTK also correlate with negative regulation of the pro-apoptotic tumor suppressor WW domain-containing oxidoreductase (Wwox) [138][105]. Also, AXL inhibition is reported to mediate apoptosis by reducing the expression of the anti-apoptotic protein MCL1 [139][106]. AXL dimerizes with and phosphorylates EGFR to promote activation of the PLCγ-PKC-mTOR signaling cascade and tumor cell survival [76][62]. Similarly, there is crosstalk between MERTK and EGFR and they are frequently co-expressed on both mtEGFR- and wtEGFR-expressing NSCLC cell lines [69,80][55][66]. In fact, MERTK stabilized the EGFR protein on the cell surface, probably by preventing EGFR internalization and degradation, as EGF-dependent EGFR turnover was reversed by inhibition of lysosomal hydrolase activity [140][107]. Further, inhibition of MERTK expression using siRNA destabilized expression of EGFR protein.3.4. Immune Regulatory Functions in the Tumor Microenvironment

3.4. Immune Regulatory Functions in the Tumor Microenvironment
MERTK is upregulated upon monocyte to macrophage differentiation [141,142,143][108][109][110]. Expression of MERTK and AXL on tumor-infiltrating macrophages polarizes them towards a pro-tumor M2-like phenotype [143,144,145,146][110][111][112][113]. M2 macrophages promote an immunosuppressive tumor microenvironment by increasing expression of wound-healing cytokines (IL-10, TGFβ, and IL-4) and decreasing pro-inflammatory cytokines (IL-12, TNFα, and IL-6) [45,143,147,148,149,150,151][31][110][114][115][116][117][118] (Figure 3A). MERTK activation negatively regulates the secretion of pro-inflammatory cytokines, such as TNFα, through suppression of NFκB activation in macrophages [45,152][31][119]. LPS challenge led to over-produced TNFα in Mertkkd mice, which lack the tyrosine kinase signaling domain, due to hyper-activation of NFκB [45,153][31][120]. Inhibition of MERTK by knockout of Mertk in mice, neutralization of TAM kinase signaling using a recombinant MERTK-Fc protein as a ligand sink or a GAS6 blocking antibody, and knockout of AXL in macrophages also impaired M2-macrophage anti-inflammatory phenotypes, decreased immunosuppressive IL-10 production, and increased pro-inflammatory IL-12 release [86,143,154,155][72][110][121][122]. These cytokine alterations lead to expansion of anti-tumor CD8+ T lymphocytes and inhibition of tumor growth and metastasis (Figure 3B). Indeed, inhibition of MERTK in the tumor microenvironment in Mertk−/− mice was sufficient to decrease tumor growth and metastasis [154][121]. MERTK-expressing dendritic cells can also regulate T cell activation directly [156][123]. Blocking MERTK on dendritic cells using anti-MERTK antibody promoted T cell proliferation, while treatment with a MERTK-Fc protein to mimic the effect of MERTK expressed on human dendritic cells suppressed naïve CD4+ T cell proliferation [156][123]. The anti-inflammatory effect of MERTK activation in macrophages and apoptotic cell-treated dendritic cells was mediated by inhibition of NFκB activation [50,157,158][36][124][125] or by induction of toll-like receptor (TLR) suppressor of cytokine signaling 1 (SOCS1) and SOCS3 [158,159,160][125][126][127] (Figure 2). Further, the MERTK ligand PROS1 also promotes resolution of inflammation by macrophages and inhibits macrophage M1 polarization to reduce anti-tumor immune response [161][128].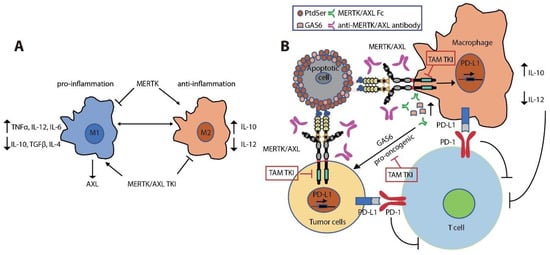
References
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500.
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565.
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139.
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203.
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246.
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334.
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394.
- Alamgeer, M.; Ganju, V.; Watkins, D.N. Novel therapeutic targets in non-small cell lung cancer. Curr. Opin. Pharmacol. 2013, 13, 394–401.
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133.
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539.
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17.
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562.
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709.
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031.
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657.
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910.
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407.
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985.
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3.
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027.
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764.
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763.
- Crosier, P.S.; Freeman, S.A.; Orlic, D.; Bodine, D.M.; Crosier, K.E. The Dtk receptor tyrosine kinase, which binds protein S, is expressed during hematopoiesis. Exp. Hematol. 1996, 24, 318–323.
- Faust, M.; Ebensperger, C.; Schulz, A.S.; Schleithoff, L.; Hameister, H.; Bartram, C.R.; Janssen, J.W. The murine ufo receptor: Molecular cloning, chromosomal localization and in situ expression analysis. Oncogene 1992, 7, 1287–1293.
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311.
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211.
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728.
- Camenisch, T.D.; Koller, B.H.; Earp, H.S.; Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999, 162, 3498–3503.
- Caraux, A.; Lu, Q.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat. Immunol. 2006, 7, 747–754.
- Rahman, Z.S.; Shao, W.H.; Khan, T.N.; Zhen, Y.; Cohen, P.L. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J. Immunol. 2010, 185, 5859–5868.
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140.
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246.
- Wallet, M.A.; Sen, P.; Flores, R.R.; Wang, Y.; Yi, Z.; Huang, Y.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; et al. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008, 205, 219–232.
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431.
- Wimmel, A.; Glitz, D.; Kraus, A.; Roeder, J.; Schuermann, M. Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur. J. Cancer 2001, 37, 2264–2274.
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860.
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259.
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219.
- Iida, S.; Miki, Y.; Suzuki, T.; Mori, K.; Saito, M.; Niikawa, H.; Kondo, T.; Yamada-Okabe, H.; Sasano, H. Activation of AXL and antitumor effects of a monoclonal antibody to AXL in lung adenocarcinoma. Anticancer Res. 2014, 34, 1821–1827.
- Qu, X.H.; Liu, J.L.; Zhong, X.W.; Li, X.I.; Zhang, Q.G. Insights into the roles of hnRNP A2/B1 and AXL in non-small cell lung cancer. Oncol. Lett. 2015, 10, 1677–1685.
- Wu, Z.; Bai, F.; Fan, L.; Pang, W.; Han, R.; Wang, J.; Liu, Y.; Yan, X.; Duan, H.; Xing, L. Coexpression of receptor tyrosine kinase AXL and EGFR in human primary lung adenocarcinomas. Hum. Pathol. 2015, 46, 1935–1944.
- Ramkumar, K.; Stewart, C.A.; Cargill, K.R.; Della Corte, C.M.; Wang, Q.; Shen, L.; Diao, L.; Cardnell, R.J.; Peng, D.H.; Rodriguez, B.L.; et al. AXL Inhibition Induces DNA Damage and Replication Stress in Non-Small Cell Lung Cancer Cells and Promotes Sensitivity to ATR Inhibitors. Mol. Cancer Res. 2021, 19, 485–497.
- Safaric Tepes, P.; Pal, D.; Lindsted, T.; Ibarra, I.; Lujambio, A.; Jimenez Sabinina, V.; Senturk, S.; Miller, M.; Korimerla, N.; Huang, J.; et al. An epigenetic switch regulates the ontogeny of AXL-positive/EGFR-TKi-resistant cells by modulating miR-335 expression. eLife 2021, 10.
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064.
- Ishikawa, M.; Sonobe, M.; Nakayama, E.; Kobayashi, M.; Kikuchi, R.; Kitamura, J.; Imamura, N.; Date, H. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann. Surg. Oncol. 2013, 20 (Suppl. 3), S467–S476.
- Melchionna, R.; Spada, S.; Di Modugno, F.; D’Andrea, D.; Di Carlo, A.; Panetta, M.; Mileo, A.M.; Sperduti, I.; Antoniani, B.; Gallo, E.; et al. The actin modulator hMENA regulates GAS6-AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020, 21, e50078.
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455.
- Sato, K.; Suda, K.; Shimizu, S.; Sakai, K.; Mizuuchi, H.; Tomizawa, K.; Takemoto, T.; Nishio, K.; Mitsudomi, T. Clinical, Pathological, and Molecular Features of Lung Adenocarcinomas with AXL Expression. PLoS ONE 2016, 11, e0154186.
- Cummings, C.T.; Linger, R.M.; Cohen, R.A.; Sather, S.; Kirkpatrick, G.D.; Davies, K.D.; DeRyckere, D.; Earp, H.S.; Graham, D.K. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget 2014, 5, 10434–10445.
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 2014, 57, 7031–7041.
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022.
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S., 3rd; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancers Expressing Wild-type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535.
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264.
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898.
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164.
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.M.; et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014, 74, 253–262.
- Wu, F.; Li, J.; Jang, C.; Wang, J.; Xiong, J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6653–6661.
- Bae, S.Y.; Hong, J.Y.; Lee, H.J.; Park, H.J.; Lee, S.K. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget 2015, 6, 10146–10160.
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546.
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253.
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308.
- Nakamichi, S.; Seike, M.; Miyanaga, A.; Chiba, M.; Zou, F.; Takahashi, A.; Ishikawa, A.; Kunugi, S.; Noro, R.; Kubota, K.; et al. Overcoming drug-tolerant cancer cell subpopulations showing AXL activation and epithelial-mesenchymal transition is critical in conquering ALK-positive lung cancer. Oncotarget 2018, 9, 27242–27255.
- Yan, D.; Huelse, J.; Parker, R.; Tan, Z.; Wang, X.; Frye, S.V.; Earp, H.S.; DeRyckere, D.; Graham, D.K. MERTK drives residual tumor growth in EGFR-mutated non-small cell lung cancer cells treated with osimertinib. Cancer Res. 2020, 80.
- Yan, D.; Tan, Z.; Wang, X.; Frye, S.V.; Earp, H.S., III; DeRyckere, D.; Graham, D.K. A novel strategy to cope with osimertinib resistance in non-small cell lung cancer by treatment with a PIM kinase inhibitor in combination with a MERTK-selective kinase inhibitor. Cancer Res. 2021, 81.
- Okura, N.; Nishioka, N.; Yamada, T.; Taniguchi, H.; Tanimura, K.; Katayama, Y.; Yoshimura, A.; Watanabe, S.; Kikuchi, T.; Shiotsu, S.; et al. ONO-7475, a Novel AXL Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2244–2256.
- Rios-Doria, J.; Favata, M.; Lasky, K.; Feldman, P.; Lo, Y.; Yang, G.; Stevens, C.; Wen, X.; Sehra, S.; Katiyar, K.; et al. A Potent and Selective Dual Inhibitor of AXL and MERTK Possesses Both Immunomodulatory and Tumor-Targeted Activity. Front. Oncol. 2020, 10, 598477.
- Wang, T.H.; Wu, C.C.; Huang, K.Y.; Leu, Y.L.; Yang, S.C.; Chen, C.L.; Chen, C.Y. Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer. Cancers 2020, 13, 111.
- Konen, J.M.; Rodriguez, B.L.; Padhye, A.; Ochieng, J.K.; Gibson, L.; Diao, L.; Fowlkes, N.W.; Fradette, J.J.; Peng, D.H.; Cardnell, R.J.; et al. Dual Inhibition of MEK and AXL Targets Tumor Cell Heterogeneity and Prevents Resistant Outgrowth Mediated by the Epithelial-to-Mesenchymal Transition in NSCLC. Cancer Res. 2021, 81, 1398–1412.
- Tirado-Gonzalez, I.; Descot, A.; Soetopo, D.; Nevmerzhitskaya, A.; Schaffer, A.; Czlonka, E.; Wachtel, C.; Tsoukala, I.; Muller, L. AXL Inhibition in Macrophages Stimulates Host-versus-Leukemia Immunity and Eradicates Naive and Treatment-Resistant Leukemia. Cancer Discov. 2021, 11.
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890.
- Kim, K.C.; Baek, S.H.; Lee, C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int. J. Oncol. 2015, 47, 2296–2303.
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219.
- Shin, D.H.; Lee, H.J.; Min, H.Y.; Choi, S.P.; Lee, M.S.; Lee, J.W.; Johnson, F.M.; Mehta, K.; Lippman, S.M.; Glisson, B.S.; et al. Combating resistance to anti-IGFR antibody by targeting the integrin beta3-Src pathway. J. Natl. Cancer Inst. 2013, 105, 1558–1570.
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Fargnoli, J.; Graf Finckenstein, F.; Gottardis, M.M.; Carboni, J.M. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010, 70, 7221–7231.
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212.
- Graham, D.K.; Bowman, G.W.; Dawson, T.L.; Stanford, W.L.; Earp, H.S.; Snodgrass, H.R. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene 1995, 10, 2349–2359.
- Ling, L.; Kung, H.J. Mitogenic signals and transforming potential of Nyk, a newly identified neural cell adhesion molecule-related receptor tyrosine kinase. Mol. Cell. Biol. 1995, 15, 6582–6592.
- Lierman, E.; Van Miegroet, H.; Beullens, E.; Cools, J. Identification of protein tyrosine kinases with oncogenic potential using a retroviral insertion mutagenesis screen. Haematologica 2009, 94, 1440–1444.
- Georgescu, M.M.; Kirsch, K.H.; Shishido, T.; Zong, C.; Hanafusa, H. Biological effects of c-Mer receptor tyrosine kinase in hematopoietic cells depend on the Grb2 binding site in the receptor and activation of NF-kappaB. Mol. Cell. Biol. 1999, 19, 1171–1181.
- Janssen, J.W.; Schulz, A.S.; Steenvoorden, A.C.; Schmidberger, M.; Strehl, S.; Ambros, P.F.; Bartram, C.R. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 1991, 6, 2113–2120.
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448.
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290.
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol. Cancer Res. 2017, 15, 45–58.
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129.
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6.
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161.
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26.
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272.
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454.
- Bansal, N.; Mishra, P.J.; Stein, M.; DiPaola, R.S.; Bertino, J.R. Axl receptor tyrosine kinase is up-regulated in metformin resistant prostate cancer cells. Oncotarget 2015, 6, 15321–15331.
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691.
- Cichon, M.A.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; Mackenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2014, 33, 4185–4192.
- Lay, J.D.; Hong, C.C.; Huang, J.S.; Yang, Y.Y.; Pao, C.Y.; Liu, C.H.; Lai, Y.P.; Lai, G.M.; Cheng, A.L.; Su, I.J.; et al. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007, 67, 3878–3887.
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene 2008, 27, 4044–4055.
- Ling, L.; Templeton, D.; Kung, H.J. Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 18355–18362.
- Weinger, J.G.; Gohari, P.; Yan, Y.; Backer, J.M.; Varnum, B.; Shafit-Zagardo, B. In brain, Axl recruits Grb2 and the p85 regulatory subunit of PI3 kinase; in vitro mutagenesis defines the requisite binding sites for downstream Akt activation. J. Neurochem. 2008, 106, 134–146.
- Braunger, J.; Schleithoff, L.; Schulz, A.S.; Kessler, H.; Lammers, R.; Ullrich, A.; Bartram, C.R.; Janssen, J.W. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997, 14, 2619–2631.
- Fridell, Y.W.; Jin, Y.; Quilliam, L.A.; Burchert, A.; McCloskey, P.; Spizz, G.; Varnum, B.; Der, C.; Liu, E.T. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 1996, 16, 135–145.
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453.
- Rogers, A.E.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2012, 31, 4171–4181.
- Besser, D.; Bromberg, J.F.; Darnell, J.E., Jr.; Hanafusa, H. A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol. Cell. Biol. 1999, 19, 1401–1409.
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005, 65, 10514–10523.
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937.
- Komurov, K.; Padron, D.; Cheng, T.; Roth, M.; Rosenblatt, K.P.; White, M.A. Comprehensive mapping of the human kinome to epidermal growth factor receptor signaling. J. Biol. Chem. 2010, 285, 21134–21142.
- Zahuczky, G.; Kristof, E.; Majai, G.; Fesus, L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS ONE 2011, 6, e21349.
- Behrens, E.M.; Gadue, P.; Gong, S.Y.; Garrett, S.; Stein, P.L.; Cohen, P.L. The mer receptor tyrosine kinase: Expression and function suggest a role in innate immunity. Eur. J. Immunol. 2003, 33, 2160–2167.
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520.
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128.
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94.
- Mahajan, N.P.; Earp, H.S. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: A mechanism for localizing guanine nucleotide-exchange factor action. J. Biol. Chem. 2003, 278, 42596–42603.
- Crittenden, M.R.; Baird, J.; Friedman, D.; Savage, T.; Uhde, L.; Alice, A.; Cottam, B.; Young, K.; Newell, P.; Nguyen, C.; et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 2016, 7, 78653–78666.
- Kodelja, V.; Muller, C.; Tenorio, S.; Schebesch, C.; Orfanos, C.E.; Goerdt, S. Differences in angiogenic potential of classically vs alternatively activated macrophages. Immunobiology 1997, 197, 478–493.
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118.
- Alciato, F.; Sainaghi, P.P.; Sola, D.; Castello, L.; Avanzi, G.C. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J. Leukoc. Biol. 2010, 87, 869–875.
- Kim, S.Y.; Lim, E.J.; Yoon, Y.S.; Ahn, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci. Rep. 2016, 6, 29673.
- Ito, K. Impact of post-translational modifications of proteins on the inflammatory process. Biochem. Soc. Trans. 2007, 35, 281–283.
- Covert, M.W.; Leung, T.H.; Gaston, J.E.; Baltimore, D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 2005, 309, 1854–1857.
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242.
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637.
- Cabezon, R.; Carrera-Silva, E.A.; Florez-Grau, G.; Errasti, A.E.; Calderon-Gomez, E.; Lozano, J.J.; Espana, C.; Ricart, E.; Panes, J.; Rothlin, C.V.; et al. MERTK as negative regulator of human T cell activation. J. Leukoc. Biol. 2015, 97, 751–760.
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S., Jr.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 2007, 109, 653–660.
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136.
- Nakagawa, R.; Naka, T.; Tsutsui, H.; Fujimoto, M.; Kimura, A.; Abe, T.; Seki, E.; Sato, S.; Takeuchi, O.; Takeda, K.; et al. SOCS-1 participates in negative regulation of LPS responses. Immunity 2002, 17, 677–687.
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076.
- Lumbroso, D.; Soboh, S.; Maimon, A.; Schif-Zuck, S.; Ariel, A.; Burstyn-Cohen, T. Macrophage-Derived Protein S Facilitates Apoptotic Polymorphonuclear Cell Clearance by Resolution Phase Macrophages and Supports Their Reprogramming. Front. Immunol. 2018, 9, 358.
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e359.
- Scutera, S.; Fraone, T.; Musso, T.; Cappello, P.; Rossi, S.; Pierobon, D.; Orinska, Z.; Paus, R.; Bulfone-Paus, S.; Giovarelli, M. Survival and migration of human dendritic cells are regulated by an IFN-alpha-inducible Axl/Gas6 pathway. J. Immunol. 2009, 183, 3004–3013.
- Sharif, M.N.; Sosic, D.; Rothlin, C.V.; Kelly, E.; Lemke, G.; Olson, E.N.; Ivashkiv, L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 2006, 203, 1891–1901.
- Yoshimura, A.; Nishinakamura, H.; Matsumura, Y.; Hanada, T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res. Ther. 2005, 7, 100–110.
- Mansell, A.; Smith, R.; Doyle, S.L.; Gray, P.; Fenner, J.E.; Crack, P.J.; Nicholson, S.E.; Hilton, D.J.; O’Neill, L.A.; Hertzog, P.J. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol. 2006, 7, 148–155.
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928.
- Du, W.; Huang, H.; Sorrelle, N.; Brekken, R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 2018, 3, e124184.
- Paolino, M.; Penninger, J.M. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97.
- Group NM-aC; Arriagada, R.; Auperin, A.; Burdett, S.; Higgins, J.P.; Johnson, D.H.; Le Chevalier, T.; Le Pechoux, C.; Parmar, M.K.; Pignon, J.P.; et al. Adjuvant chemotherapy, with or without postoperative radiotherapy, in operable non-small-cell lung cancer: Two meta-analyses of individual patient data. Lancet 2010, 375, 1267–1277.
- Lee, H.J.; Pham, P.C.; Pei, H.; Lim, B.; Hyun, S.Y.; Baek, B.; Kim, B.; Kim, Y.; Kim, M.H.; Kang, N.W.; et al. Development of the phenylpyrazolopyrimidine-based, insulin-like growth factor receptor/Src/AXL-targeting small molecule kinase inhibitor. Theranostics 2021, 11, 1918–1936.
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941.
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM tyrosine kinase inhibitor BMS-777607 enhances anti-PD-1 mAb efficacy in a murine model of triple-negative breast cancer. Cancer Res. 2019, 79, 2669–2683.
- Tsukita, Y.; Fujino, N.; Miyauchi, E.; Saito, R.; Fujishima, F.; Itakura, K.; Kyogoku, Y.; Okutomo, K.; Yamada, M.; Okazaki, T.; et al. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol. Cancer 2019, 18, 24.
- Terry, S.; Dalban, C.; Rioux Leclercq, N.; Adam, J.; Meylan, M.; Buart, S.; Bougouin, A.; Lespagnol, A.; Dugay, F.; Colina Moreno, I.; et al. Association of AXL and PD-L1 expression with clinical outcomes in patients with advanced renal cell carcinoma treated with PD-1 blockade. Clin. Cancer Res. 2021.
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802.
- Grabiec, A.M.; Goenka, A.; Fife, M.E.; Fujimori, T.; Hussell, T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur. J. Immunol. 2018, 48, 855–860.
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007, 178, 5635–5642.
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618.
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791.
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738.
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52.
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830.
- Bianchi, B.R.; Lynch, K.J.; Touma, E.; Niforatos, W.; Burgard, E.C.; Alexander, K.M.; Park, H.S.; Yu, H.; Metzger, R.; Kowaluk, E.; et al. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur. J. Pharmacol. 1999, 376, 127–138.
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642.
- Tanvetyanon, T.; Gray, J.E.; Antonia, S.J. PD-1 checkpoint blockade alone or combined PD-1 and CTLA-4 blockade as immunotherapy for lung cancer? Expert Opin. Biol. Ther. 2017, 17, 305–312.
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833.