IL-6 is a pleiotropic cytokine showing both pro- and anti-inflammatory roles.
- IL-6 signaling
- membrane IL-6 receptor
- soluble IL-6 receptor
- colorectal cancer
- polyposis syndromes
- IL-6-targeting therapy
1. Introduction
CRC is one of the most frequent cancers worldwide and one of the main causes of cancer-related mortality [1]. The onset of CRC is associated with genetic and environmental factors. In hereditary or familial colorectal cancer, genetic predisposition plays a crucial role in disease onset, and germline inactivating mutations in specific oncogenes or tumor-suppressor genes involved in the physiological turnover of colorectal mucosae could predispose a person to hereditary colorectal cancer syndrome. Furthermore, minor variants in the same genes responsible for hereditary cancers cause familial cancers. However, environmental factors represent the main cause of cancer onset for sporadic forms [2,3][2][3]. About 30% of all CRCs are hereditary or familial, while about 75% are sporadic [4]. The first-degree relatives of patients with CRC have a three-fold greater risk of CRC than individuals without familial predisposition [5].
During cancer progression, healthy mucosa accumulates genetic or epigenetic alterations in oncogenes or tumor-suppressor genes, evolving into hyperproliferative mucosa and, later, into early, intermediate, and late adenomas, which, in turn, give rise to in situ carcinomas. Eventually, the accumulation of altering mutations in genes involved in cell–cell adhesion, epithelial-to-mesenchymal transition, and extracellular matrix degradation confer to the cancer cells the ability for distant colonization, generating metastases. This well-known sequence, named the adenoma–carcinoma sequence, is also determined by growth factors and cytokines [6].
Lynch syndrome (LS), also known as non-polyposis colorectal syndrome (HNPCC), is a hereditary disease that accounts for about 1–6% of CRCs. LS predisposes one to CRC onset at a younger age than the general population, usually before 50 years [7[7][8][9],8,9], and to an increase in cancer risk in several regions such as the endometrium, stomach, liver, kidney, and brain, as well as an increased risk of certain types of skin cancer. Germline inactivating mutations in mismatch repair (MMR) genes, such as MLH1, MSH2, MSH6, and PMS2, have been associated with LS.
Polyposis syndromes are a heterogeneous group of hereditary syndromes predisposing to colorectal cancers, characterized by the onset of several polyposes of the colon and rectum, which are classified as adenomatous polyposis and hamartomatous polyposis syndromes. Adenomatous polyposis syndromes include familial adenomatous polyposis (FAP), attenuated FAP (AFAP), and MUTYH-associated polyposis (MAP); hamartomatous polyposis syndromes consist of PTEN hamartoma tumor syndrome (PHTS), juvenile polyposis syndrome (JPS), and Peutz–Jeghers syndrome (PJS). While MAP is an autosomal recessive condition, the other polyposis syndromes, both adenomatous and hamartomatous, are inherited in an autosomal dominant manner [1,6][1][6]. FAP is an autosomal dominant condition characterized by the development of hundreds to thousands of adenomatous polyps in the gastrointestinal tract, usually within the second decade of life, which, if untreated, inevitably evolve into carcinomas [10]. Phenotypic variability has been described to be associated with specific gene alterations or the localization of the pathogenic variant on the gene, including attenuated, classic, and aggressive phenotypes, characterized by different numbers of polyps and onset ages. Adenomatous polyposis patients can also develop extraintestinal manifestations, including congenital hypertrophy of the retinal pigment epithelium (CHRPE), polyposis of the upper intestinal tract, desmoid tumors, thyroid tumors, and hepatoblastoma [11]. Biallelic germline mutations of the MutY DNA glycosylase (MUTYH) gene and monoallelic mutations of the adenomatous polyposis coli (APC) gene both cause FAP syndromes. However, mutations in MUTYH result in a less severe phenotype than mutations in APC, characterized by the onset of fewer than 100 adenomas and a late age of disease manifestation [12,13][12][13]. Pathogenic germline variants in other genes, such as NTHL1, POLE, and POLD1, are also described in polyposis patients [1]. Although many genes are now known to be associated with the onset of polyposis syndromes when altered, polyposis patients often remain without an identified pathogenic genomic variant [14]. The main genes that, when altered, are involved in the hamartomatous polyposis syndromes are the STK11 serine/threonine kinase, associated with PJS syndrome, and the tumor-suppressor gene PTEN, associated with PHTS syndrome. Other genes suggested to be involved in hamartomatous syndromes are BMPR1A, SDHB, SDHD, SMAD4, AKT1, ENG, and PIK3CA [1,15,16][1][15][16].
It is becoming evident that there is a role for interleukin-6 in inflammation and carcinogenesis, through its downstream transcription factors, such as signal transducer and transcription 3 (STAT3), which stimulates cancer cell proliferation and migration. It has been suggested that the main sources of interleukin-6 (IL-6) production during colorectal cancer progression are the tumor-associated macrophages, mesenchymal stem cells, or colon cancer-associated fibroblasts [17,18][17][18]. These data are corroborated by the observations that both membrane and soluble forms of interleukin-6 receptor (IL-6R) are upregulated at the blood serum level in colon cancer patients, and this increase correlates with the cancer size, suggesting a role for IL-6R in colorectal cancer progression. In agreement with this observation, the loss of APC, which is a gene considered to be a gatekeeper of intestinal epithelial turnover, has been described to be associated with the upregulation of IL-6 signaling, which, in turn, activates families of proteins that are often upregulated in CRC, such as the Src family kinases (SFKs), YAP, and STAT3 [19].
2. IL-6 Signaling
IL-6 is a pleiotropic cytokine taking on pro- or anti-inflammatory roles, produced by many cell types, such as stromal, hematopoietic, epithelial, and muscle cells. It plays a role in various biological mechanisms, such as immune responses, cell survival, apoptosis, and proliferation [20[20][21][22][23],21,22,23], by acting in concert with other factors, including heparin-binding epithelial growth factor and hepatocyte growth factor [24,25,26,27,28][24][25][26][27][28]. IL-6 also regulates the proliferation of intestinal epithelial cells [24] and is involved in the differentiation of various cells, including mast cells and cardiomyocytes [29]. Furthermore, its anti- and pro-inflammatory properties make it able to modulate the responses to several diseases, including immune diseases (e.g., rheumatoid arthritis); chronic inflammatory diseases; and cancers, such as prostatic and bladder cancers, neurological cancers, and B-cell malignancies [25,30,31,32,33][25][30][31][32][33]. However, it is also implicated in Alzheimer’s disease, myocardial infarction, Paget’s disease, and osteoporosis [25].
There are two different ways in which IL-6 signaling can be activated. The first is classic signaling, which involves binding between IL-6 and its membrane receptors, the IL-6R (gp80, α-chain, or CD126) [25], on the surface of target cells. Alternatively, IL-6 binds to a soluble interleukin-6 receptor (sIL-R) (or gp55) in a process called “IL-6 trans-signaling”. As shown in Figure 1, transmembrane metalloproteases are able to cleave membrane IL-6R (mIL-6R) and promote the release of the sIL-6R from membrane receptors localized on the plasma membrane of IL-6 target cells, activating IL-6 trans-signaling [34]. Soluble IL-6R can also be produced by a transcriptional mechanism that generates an alternatively spliced mRNA isoform of the receptor without the region encoding the transmembrane domain, yielding a protein that differs at its COOH terminus by 14 amino acid residues [35,36,37][35][36][37]. This mechanism allows the activation of IL-6 signaling even without IL-6R, when the level of sIL-6R circulating is very high, as observed in several pathological conditions.
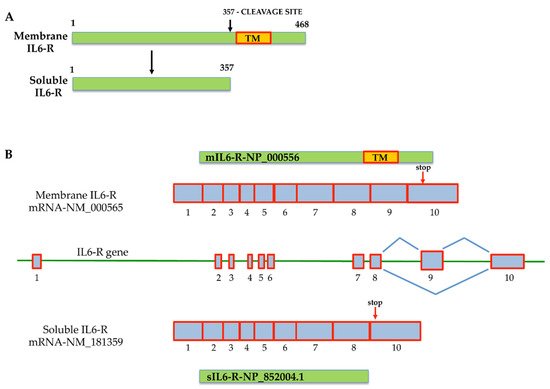
Figure 1. Mechanisms that generate membrane interleukin-6 receptor (mIL-6R) and soluble interleukin-6 receptor (sIL-6R). (A) Generation of the sIL-6R protein isoform by metalloprotease cleavage of mIL-6R that removes the transmembrane protein domain; (B) generation of the two interleukin-6 receptors (IL-6R) by alternative splicing. The organization of the two alternative splicing isoforms is described. The sIL6-R mRNA is generated by the loss of exon 9, encoding for the transmembrane protein domain (TM). The mIL-6R messenger isoform retains exon 9. The scheme is based on data from NCBI, and the accession numbers of corresponding sequences are reported. Red arrows indicate the stop codon position on exon 10 in both messengers.
Scientific evidence suggests that IL-6 trans-signaling mainly acts in a pro-inflammatory manner, promoting neoplastic transformation, whereas IL-6 classic signaling mainly takes place in regenerative or anti-inflammatory processes (Figure 1) [38]. As shown in Figure 2, IL-6 binding to its receptor (IL-6R or sIL6-R) induces the IL-6/IL-6R complex’s interaction with the ubiquitously expressed gp130 IL-6 transducer (β-chain and CD130). This, in turn, results in gp130 dimerization and phosphorylation and the activation of downstream targets, such as receptor-associated kinases (JAK1, JAK2, and Tyk2), which are eventually responsible for cell proliferation and tumor progression [25]. JAK phosphorylation results in the phosphorylation and activation of the transcription factor signal transducer and activator of transcription 3 (STAT3), which represents a crucial step for cancer transformation and progression through the activation of specific target genes typically involved in neoplastic transformation. Indeed, genes involved in cell survival (Bcl2 and survivin); cell proliferation (c-Myc, cyclin D1, and cyclin B); angiogenesis (HIF-1alpha and VEGF); extracellular matrix degradation (MMP2 and MMP9); cell adhesion (ICAM-1); and inflammation (IL-6, IL-17, IL-23, and Cox2) are among STAT3’s target genes (Figure 1) [38].
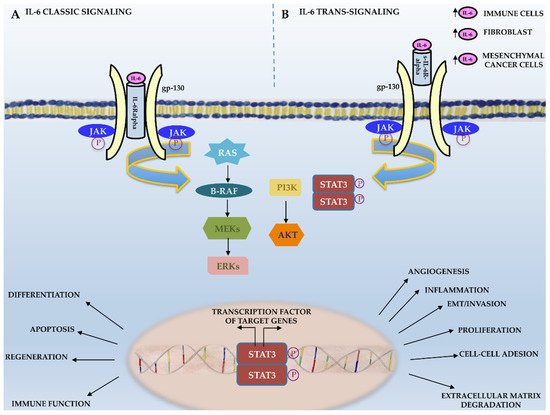
Figure 2. IL-6 signaling. Schematic representation of IL-6 classic and trans-signaling.
3. Role of the IL-6 Signaling in CRC
Since IL-6 is able to activate pro- and anti-inflammatory cell responses, it also shows pro- or anti-neoplastic activity, in a cell-specific manner, that is associated with the presence or loss of IL-6R [25]. Different mechanisms are involved in IL-6’s antitumor activity, such as the promotion of macrophage and lymphokine-activated killer cell antitumor activities, as well as an increase in neutrophils’ cytotoxic effects on tumor cells. IL-6 also sustains cancer cell lysis through the activation of C-reactive protein (CRP) and its binding to the phospholipids of cancer cells, which, in turn, activates the component 1q of the complement system. However, IL-6 signaling often induces cancer progression in both autocrine and paracrine manners (Figure 3). In an autocrine manner, cancer cells upregulate cytokines or their receptors [25], while it is now evident that, in several cancer cells—mainly in solid tumors—cytokines act in a paracrine manner [39]. In agreement with these observations, IL-6, as well as IL-6R, and other proteins and cytokines of the acute phase response such as IL-1 and TNF-alpha are upregulated during tumor progression, and they are elevated in the serum in the late tumor stage, which correlates with the disease severity and outcomes [40,41,42,43][40][41][42][43]. The main sources of IL-6 in CRC are tumor-associated macrophages, mesenchymal stem cells, and IL-6 released by colon cancer-associated fibroblasts [44]. Experimental and clinical studies demonstrated a clear association of sporadic CRC and inflammation-associated colorectal cancer with IL-6 signaling, although the specific mechanism through which IL-6 plays a role during CRC onset and progression has not been completely clarified.
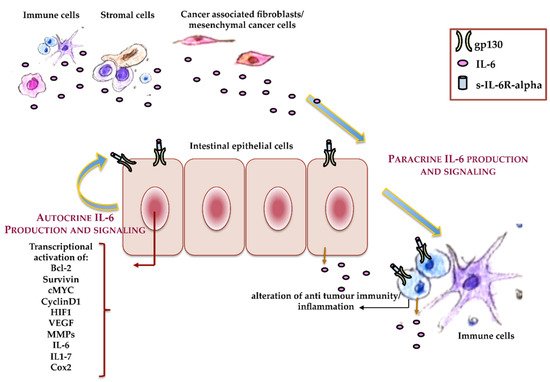
Figure 3. Autocrine and paracrine IL-6. Schematic representation of the roles that autocrine and paracrine IL-6 play during colorectal cancer (CRC) onset and progression.
In vitro studies have demonstrated that IL-6 promotes the growth of epithelial colon cancer cells [38,45,46][38][45][46]. Furthermore, scientific data show the upregulation of serum IL-6 in CRC, which correlates with the tumor size [47,48][47][48] but, also, with poor prognosis in metastatic colon cancer patients [49,50][49][50] and treatment-refractory carcinomas [51,52][51][52]. These observations suggest that the blood serum concentration of IL-6 could be considered to be a diagnostic and prognostic biomarker in colorectal cancer patients that correlates with relapse-free survival and recurrence [44,45,47,53,54][44][45][47][53][54]. Furthermore, blood serum IL-6 is upregulated in CRC patients, and tissue IL-6 expression is higher in CRC than in healthy colon mucosa. This increase, again, positively correlates with the tumor TNM stage, invasion, and lymph node metastases and risk of relapse [44,55,56[44][55][56][57][58],57,58], while a negative correlation has been observed with tumor histological differentiation. These findings support the observation that a high tumor tissue IL-6 concentration is associated with a poor prognosis in patients with CRC, and it might be a useful predictive marker. It has also been suggested to be a potential therapeutic target in CRC [38,44,57][38][44][57]. Recent literature data have shown that a low level of cancer tissue IL-6 improves the disease-free survival as compared with tumor counterpart tissues expressing higher IL-6. It has been suggested that IL-6 could represent a marker for evaluating the immune status of both PD-L1-negative and PD-L1-positive colorectal cancer patients, which could provide useful information for determining the therapeutic strategies [49]. In agreement with this hypothesis, a previous report demonstrated that the inhibition of IL-6 enhanced the efficacy of anti-PD-L1 treatment in murine models of pancreatic cancer [59].
There are several mechanisms by which IL-6 drives cancer progression. Holmer et al. performed in vitro studies and showed a relationship between the expression of IL-6 and IL-6R in a colon carcinoma cell line and the upregulation of the carcinoembryonic antigen (CEA). They observed that IL-6 classic signaling induced only a weak upregulation of CEACAM5 and CEACAM6; however, IL-6 trans-signaling was able to strongly upregulate the CEA antigens in a STAT3-phosphorylation-dependent manner [60]. This finding is in agreement with the observation that intestinal epithelial cells usually do not express membrane IL-6R (CD126) [38]. It has also been demonstrated that vascular endothelial growth factor receptor 2 (VEGFR2) is under IL-6 control in intestinal epithelial cells. Indeed, during inflammation and tumor conditions, cancer stromal fibroblasts upregulate IL-6 release, which, in turn, induces tumor angiogenesis. Interestingly, IL-6R neutralization negatively regulates angiogenesis and tumor growth through IL-6 signaling downregulation, supporting the hypothesis that considers IL-6 signaling proteins to be useful targets in CRC therapy [17,61][17][61] and in several other human diseases [49]. The FRA1 protein is among the downstream targets of the IL-6/STAT3 complex. It is a member of the FOS family transcription factors encoded by the FOS ligand 1 (FOSL1) gene that is involved in CRC progression and aggressiveness through EMT activation [62,63][62][63]. IL-6-mediated STAT3 activation upregulates HDAC6 deacetylase, which induces FRA1 Lys-116 deacetylation and FRA1 transcriptional activation, which, in turn, activates the expression of NANOG and other stem cell and EMT-specific proteins, leading to CRC progression, aggressiveness, and metastatic transformation. In agreement with these observations, FRA1 has been found to be highly expressed in multiple cancers and plays a crucial role in cancer transformation, cancer cell motility, cancer cell stemness features, and drug resistance [44,64,65,66,67][44][64][65][66][67]. Furthermore, it has recently been suggested that IL-6 could promote colorectal cancer progression through the induction of EMT, metastatic cell spread [68[68][69],69], angiogenesis, self-renewal, and drug resistance [35,36,37][35][36][37]. It also could negatively affect the host’s antitumor immunity, altering the activity of dendritic cells and cytotoxic T cells that infiltrate tumor microenvironments [49].
CRC progression is also under the influence of tumor-infiltrating lymphocytes (TILs), whose increase is associated with better cancer prognosis [70]. TGF-beta production by TILs downregulates the expression of T-cell IL-6 and prevents cancer progression. Indeed, the suppression of TGF-beta signaling induces IL-6-dependent colorectal cancer cell growth. Interestingly, trans-signaling represents the principal active mechanism of IL-6-mediated signal transduction in CRC cells. A large subgroup of CRC presents a shift from the membrane-localized IL-6 receptor to the soluble isoform, which is known to be involved in the adherence of CRC cells to the vascular endothelium, inducing metastasis [71]. These findings, again, suggest that sIL6-R and other proteins involved in trans-signaling could represent a good target for CRC therapy, since they are involved in all the stages of CRC development [45]. Recent studies have shown that IL-6 modulates the immune status of the tumor microenvironment in a manner that facilitates the metastatic colonization of colon cancer cells through the alteration of antitumor effector cells [49]. Furthermore, pro-inflammatory cytokines, such as IL-6 and IL-1, induce the activation of the inducible form of prostaglandin H synthase, the COX2 enzyme, providing a scientific explanation for the observed CRC-preventive effects of nonsteroid anti-inflammatory drugs [72]. Il-6 trans-signaling, together with STAT3 activation, is also involved in shifting MSH3 from the nucleus to the cytosol, inhibiting its binding with other mismatch repair proteins. MSH3 is a component of the post-replicative DNA mismatch repair system (MMR) that forms the MutS beta complex by heterodimerization with MSH2. MutS beta, which recognizes insertion–deletion loops larger than 13 nucleotides, binds to DNA mismatches, thereby initiating DNA repair. MSH3 alterations are associated with elevated microsatellite alterations at selected tetranucleotide repeats (EMAST), the most common DNA mismatch repair defect in colorectal cancers, observed in approximately 60% of analyzed cancers [73]. In this context, IL-6 signaling drives the compartmental translocation of MSH3 and the EMAST cancer phenotype.
(References would be added automatically after the entry is online)
References
- Turano, M.; Delrio, P.; Rega, D.; Cammarota, F.; Polverino, A.; Duraturo, F.; Izzo, P.; De Rosa, M. Promising Colorectal Cancer Biomarkers for Precisition Prevention and Therapy. Cancers 2019, 11, 1932.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386.
- Strisciuglio, C.; Auricchio, R.; Martinelli, M.; Staiano, A.; Giugliano, F.P.; Andreozzi, M.; De Rosa, M.; Giannetti, E.; Gianfrani, C.; Izzo, P.; et al. Autophagy genes variants and paediatric Crohn’s disease phenotype: A single-centre experience. Dig. Liver Dis. 2014, 46, 512–517.
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and Familial Colon Cancer. Gastroenterology 2010, 138, 2044–2058.
- Karsa, L.; Lignini, T.; Patnick, J.; Lambert, R.; Sauvaget, C. The dimensions of the CRC problem. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 381–396.
- De Rosa, M.; Rega, D.; Costabile, V.; Duraturo, F.; Niglio, A.; Izzo, P.; Pace, U.; DelRio, P. The biological complexity of colorectal cancer: Insights into biomarkers for early detection and personalized care. Ther. Adv. Gastroenterol. 2016, 9, 861–886.
- Duraturo, F.; Liccardo, R.; Cavallo, A.; De Rosa, M.; Grosso, M.; Izzo, P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: Probability of synergistic effects. Int. J. Cancer 2011, 129, 1643–1650.
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol. Lett. 2019, 17, 3048–3054.
- Samowitz, W.S.; Curtin, K.; Lin, H.H.; Robertson, M.A.; Schaffer, D.; Nichols, M.; Gruenthal, K.; Leppert, M.F.; Slattery, M.L. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology 2001, 121, 830–838.
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; DelRio, P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096.
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733.
- De Rosa, M.; Galatola, M.; Borriello, S.; Duraturo, F.; Masone, S.; Izzo, P. Implication of Adenomatous Polyposis Coli and MUTYH Mutations in Familial Colorectal Polyposis. Dis. Colon Rectum 2009, 52, 268–274.
- Dodaro, C.; Grifasi, C.; Florio, J.; Santangelo, M.L.; Duraturo, F.; De Rosa, M.; Izzo, P.; Renda, A. The role of mutation analysis of the APC gene in the management of FAP patients. A controversial issue. Annali Italiani Chirurgia 2016, 87, 321–325.
- Herzig, D.O.; Buie, W.D.; Weiser, M.R.; You, Y.N.; Rafferty, J.F.; Feingold, D.; Steele, S.R. Clinical Practice Guidelines for the Surgical Treatment of Patients with Lynch Syndrome. Dis. Colon Rectum 2017, 60, 137–143.
- Zbuk, K.M.; Eng, C. Hamartomatous polyposis syndromes. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 492–502.
- Paparo, L.; Rossi, G.B.; DelRio, P.; Rega, D.; Duraturo, F.; Liccardo, R.; Debellis, M.; Izzo, P.; De Rosa, M. Differential expression of PTEN gene correlates with phenotypic heterogeneity in three cases of patients showing clinical manifestations of PTEN hamartoma tumour syndrome. Hered. Cancer Clin. Pract. 2013, 11, 8.
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tu-mour-stroma interaction. Br. J. Cancer 2014, 110, 469–478.
- Ullman, T.A.; Itzkowitz, S.H. Intestinal Inflammation and Cancer. Gastroenterology 2011, 140, 1807–1816.e1.
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A Key Regulator of Colorectal Cancer Development. Int. J. Biol. Sci. 2012, 8, 1248–1253.
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113.
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 2014, 26, 2–12.
- Rizzo, A.; Pallone, F.; Monteleone, G.; Fantini, M.C. Intestinal inflammation and colorectal cancer: A double-edged sword? World J. Gastroenterol. 2011, 17, 3092–3100.
- Sommer, J.; Engelowski, E.; Baran, P.; Garbers, C.; Floss, R.M.; Scheller, J. Interleukin-6, but not the interleukin-6 receptor plays a role in recovery from dextran sodium sulfate-induced colitis. Int. J. Mol. Med. 2014, 34, 651–660.
- Sido, A.; Radhakrishnan, S.; Kim, S.W.; Eriksson, E.; Shen, F.; Li, Q.; Bhat, V.; Reddivari, L.; Vanamala, J.K. A food-based approach that targets interleukin-6, a key regulator of chronic intestinal inflammation and colon carcinogenesis. J. Nutr. Biochem. 2017, 43, 11–17.
- Trikha, M.; Corringham, R.; Klein, B.; Rossi, J.-F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: A review of the rationale and clinical evidence. Clin. Cancer Res. 2003, 9, 4653–4665.
- Grant, S.L.; Hammacher, A.; Douglas, A.M.; Goss, A.G.; Mansfield, R.K.; Heath, J.K.; Begley, C.G. An unexpected biochemical and functional interaction between gp130 and the EGF receptor family in breast cancer cells. Oncogene 2002, 21, 460–474.
- Badache, A.; Hynes, E.N. Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of T47D breast cancer cells. Cancer Res. 2001, 61, 383–391.
- Wang, Y.D.; De Vos, J.; Jourdan, M.; Couderc, G.; Lu, Z.-Y.; Rossi, J.-F.; Klein, B. Cooperation between heparin-binding EGF-like growth factor and interleukin-6 in promoting the growth of human myeloma cells. Oncogene 2002, 21, 2584–2592.
- Tsuruda, T.; Jougasaki, M.; Boerrigter, G.; Huntley, B.K.; Chen, H.H.; D’Assoro, A.B.; Lee, S.C.; Larsen, A.M.; Cataliotti, A.; Burnett, J.C., Jr. Cardiotrophin-1 stimulation of cardiac fibroblast growth: Roles for glycoprotein 130/leukemia inhibitory factor receptor and the endothelin type A receptor. Circ. Res. 2002, 90, 128–134.
- Grivennikov, I.S.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108.
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S.; et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102.
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related ex-pression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235.
- Atreya, R.; Neurath, M.F. Involvement of IL-6 in the Pathogenesis of Inflammatory Bowel Disease and Colon Cancer. Clin. Rev. Allergy Immunol. 2005, 28, 187–196.
- Wolfsberg, T.G.; White, J.M. ADAMs in Fertilization and Development. Dev. Biol. 1996, 180, 389–401.
- Mülberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480.
- Horiuchi, S.; Koyanagi, Y.; Zhou, Y.; Miyamoto, H.; Yamamoto, M.; Yamamoto, N. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur. J. Immunol. 1994, 24, 1945–1948.
- Diamant, M.; Rieneck, K.; Mechti, N.; Zhang, X.-G.; Svenson, M.; Bendtzen, K.; Klein, B. Cloning and expression of an alternatively spliced mRNA encoding a soluble form of the human interleukin-6 signal transducer gp1301. FEBS Lett. 1997, 412, 379–384.
- Rose-John, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247.
- Novick, D.; Shulman, L.M.; Chen, L.; Revel, M. Enhancement of interleukin 6 cytostatic effect on human breast carcinoma cells by soluble IL-6 receptor from urine and reversion by monoclonal antibody. Cytokine 1992, 4, 6–11.
- Benoy, I.; Salgado, R.; Colpaert, C.; Weytjens, R.; Vermeulen, P.B.; Dirix, L.Y. Serum Interleukin 6, Plasma VEGF, Serum VEGF, and VEGF Platelet Load in Breast Cancer Patients. Clin. Breast Cancer 2002, 2, 311–315.
- De Vita, F.; Romano, C.; Orditura, M.; Galizia, G.; Martinelli, E.; Lieto, E.; Catalano, G. Interleukin-6 Serum Level Correlates with Survival in Advanced Gastrointestinal Cancer Patients but Is Not an Independent Prognostic Indicator. J. Interf. Cytokine Res. 2001, 21, 45–52.
- Jones, S.A.; Horiuchi, S.; Topley, N.; Yamamoto, N.; Fuller, G.M. The soluble interleukin 6 receptor: Mechanisms of production and implications in disease. FASEB J. 2000, 15, 43–58.
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706.
- Olsen, J.; Kirkeby, L.T.; Olsen, J.; Eiholm, S.; Jess, P.; Gögenur, I.; Troelsen, J.T. High interleukin-6 mRNA expression is a predictor of relapse in colon cancer. Anticancer Res. 2015, 35, 2235–2240.
- Becker, C.; Fantini, M.; Schramm, C.; Lehr, H.; Wirtz, S.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; Galle, P.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL–6 trans-signaling. Zeitschrift Gastroenterologie 2004, 42, 491–501.
- Schneider, M.R.; Hoeflich, A.; Fischer, J.R.; Wolf, E.; Sordat, B.; Lahm, H. Interleukin-6 stimulates clonogenic growth of primary and metastatic human colon carcinoma cells. Cancer Lett. 2000, 151, 31–38.
- Chung, Y.-C.; Chang, Y.-F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226.
- Galizia, G.; Orditura, M.; Romano, C.; Lieto, E.; Castellano, P.; Pelosio, L.; Imperatore, V.; Catalano, G.; Pignatelli, C.; De Vita, F. Prognostic Significance of Circulating IL-10 and IL-6 Serum Levels in Colon Cancer Patients Undergoing Surgery. Clin. Immunol. 2002, 102, 169–178.
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer Cells. Cancer Immunol. Res. 2019, 7, 1944–1957.
- Thomsen, M.; Kersten, C.; Sorbye, H.; Skovlund, E.; Glimelius, B.; Pfeiffer, P.; Johansen, J.S.; Kure, E.H.; Ikdahl, T.; Tveit, K.M.; et al. Interleukin-6 and C-reactive protein as prognostic biomarkers in meta- static colorectal cancer. Oncotarget 2016, 7, 75013–75022.
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069.
- Makuuchi, Y.; Honda, K.; Osaka, Y.; Kato, K.; Kojima, T.; Daiko, H.; Igaki, H.; Ito, Y.; Hoshino, S.; Tachibana, S.; et al. Soluble in-terleukin-6 receptor is a serum biomarker for the response of esophageal carcinoma to neoadjuvant chemoradiotherapy. Cancer Sci. 2013, 104, 1045–1051.
- Mager, L.F.; Wasmer, M.H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96.
- Rossi, J.-F.; Lu, Z.-Y.; Jourdan, M.; Klein, B. Interleukin-6 as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1248–1257.
- Kim, S.; Keku, T.O.; Martin, C.; Galanko, J.; Woosley, J.T.; Schroeder, J.C.; Satia, J.A.; Halabi, S.; Sandler, R.S. Circulating Levels of Inflammatory Cytokines and Risk of Colorectal Adenomas. Cancer Res. 2008, 68, 323–328.
- Bobe, G.; Albert, P.S.; Sansbury, L.B.; Lanza, E.; Schatzkin, A.; Colburn, N.H.; Cross, A.J. Interleukin-6 as a Potential Indicator for Prevention of High-Risk Adenoma Recurrence by Dietary Flavonols in the Polyp Prevention Trial. Cancer Prev. Res. 2010, 3, 764–775.
- Zeng, J.; Tang, Z.-H.; Liu, S.; Guo, S.-S. Clinicopathological significance of overexpression of interleukin-6 in colorectal cancer. World J. Gastroenterol. 2017, 23, 1780–1786.
- Müllberg, J.; Oberthür, W.; Lottspeich, F.; Mehl, E.; Dittrich, E.; Graeve, L.; Heinrich, P.C.; Rose-John, S. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J. Immunol. 1994, 152, 4958–4968.
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332.
- Holmer, R.; Wätzig, G.H.; Tiwari, S.; Rose-John, S.; Kalthoff, H. Interleukin-6 trans-signaling increases the expression of carci-noembryonic antigen-related cell adhesion molecules 5 and 6 in colorectal cancer cells. BMC Cancer 2015, 15, 975.
- Shao, J.; Sheng, G.G.; Mifflin, R.C.; Powell, D.W.; Sheng, H. Roles of Myofibroblasts in Prostaglandin E2–Stimulated Intestinal Epithelial Proliferation and Angiogenesis. Cancer Res. 2006, 66, 846–855.
- Wang, T.; Song, P.; Zhong, T.; Wang, X.; Xiang, X.; Liu, Q.; Chen, H.; Xia, T.; Liu, H.; Niu, Y.; et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene 2019, 38, 4932–4947.
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial–mesenchymal transition. Carcinog. 2015, 36, 459–468.
- Kakumoto, K.; Sasai, K.; Sukezane, T.; Oneyama, C.; Ishimaru, S.; Shibutani, K.; Mizushima, H.; Mekada, E.; Hanafusa, H.; Akagi, T. FRA1 is a determinant for the difference in RAS-induced transformation between human and rat fibroblasts. Proc. Natl. Acad. Sci. USA 2006, 103, 5490–5495.
- Sayan, E.A.; Stanford, R.; Vickery, R.C.; Grigorenko, E.L.; Diesch, J.; Kulbicki, K.; Edwards, R.B.; Pal, R.P.; Greaves, P.; Jarielencontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2011, 31, 1493–1503.
- Hong, A.; Moriceau, G.; Sun, L.; Lomeli, S.; Piva, M.; Damoiseaux, R.; Holmen, S.L.; Sharpless, N.E.; Hugo, W.; Lo, R.S. Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov. 2018, 8, 74–93.
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364.
- Lee, S.O.; Yang, X.; Duan, S.; Tsai, Y.; Strojny, L.R.; Keng, P.; Chen, Y. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget 2016, 7, 6626–6638.
- Albino, D.; Civenni, G.; Rossi, S.; Mitra, A.; Catapano, C.V.; Carbone, G.M. The ETS factor ESE3/EHF represses IL-6 preventing STAT3 activation and expansion of the prostate cancer stem-like compartment. Oncotarget 2016, 7, 76756–76768.
- Funada, Y.; Noguchi, T.; Kikuchi, R.; Takeno, S.; Uchida, Y.; Gabbert, H.E. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncol. Rep. 2003, 10, 309–313.
- Dowdall, J.; Winter, D.; Andrews, E.; Laug, W.; Wang, J.; Redmond, H. Soluble Interleukin 6 Receptor (sIL-6R) Mediates Colonic Tumor Cell Adherence to the Vascular Endothelium: A Mechanism for Metastatic Initiation? J. Surg. Res. 2002, 107, 1–6.
- Maihöfner, C.; Charalambous, M.P.; Bhambra, U.; Lightfoot, T.; Geisslinger, G.; Gooderham, N.J.; Colorectal Cancer Group. Ex-pression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis 2003, 24, 665–671.
- Tseng-Rogenski, S.S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 Alters Localization of hMSH3, Leading to DNA Mismatch Repair Defects in Colorectal Cancer Cells. Gastroenterology 2015, 148, 579–589.