Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rodrigo Naves and Version 2 by Conner Chen.
Mitochondria are vital organelles in eukaryotic cells that control diverse physiological processes related to energy production, calcium homeostasis, the generation of reactive oxygen species, and cell death. Several studies have demonstrated that structural and functional mitochondrial disturbances are involved in the development of different neuroinflammatory (NI) and neurodegenerative (ND) diseases (NI&NDDs) such as multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis.
- mitochondria
- neuroinflammatory diseases
- neurodegenerative diseases
1. Introduction
Mitochondria are vital organelles in eukaryotic cells that control diverse physiologi-cal processes related to the production of energy and also cellular processes such as cell death, calcium homeostasis, and the generation and modulation of reactive oxygen spe-cies (ROS) levels [1][2][3][1,2,3]. Mitochondria are highly dynamic and regulated by a fine balance between biogenesis and the degradation of defective organelles [4][5][4,5]. The shape, distribution, and size of mitochondria are controlled by coordinated cycles of fission and fusion known as mitochondrial dynamics [6], whereas damaged mitochondria are selectively removed by mitophagy. Biogenesis, mitochondrial dynamics, and clearance are crucial for the functional state of mitochondria. Abnormalities or an imbalance affecting these events may have detrimental effects on mitochondria biology and cell viability [7][8][7,8]. Diverse studies performed in cell cultures, animal models and patients have shown that disturbances in mitochondrial structure and function are involved in neurodegeneration leading to motor and cognitive deficits in neuroinflammatory (NI) and neurodegenerative (ND) diseases (NI&NDDs) such as multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS). Indeed, a common hallmark of different NI&NDDs is a bioenergetic deficit resulting from mitochondrial dysfunction. Furthermore, the impaired function of mitochondria increases ROS production and oxidative stress exacerbating mitochondrial damage and the progression of neurodegeneration [9][10][9,10]. In addition, structural and functional altera-tions in mitochondria are also associated with the pathological accumulation of protein aggregates in NI&NDDs. Remarkably, the restoration of mitochondrial function leads to cell damage recovery and the amelioration of clinical symptoms in cellular and animal models of NI&NDDs [11][12][13][14][15][16][11,12,13,14,15,16]. Therefore, strategies designed to restore mitochondrial homeostasis represent potential therapies for NI&NDDs and should consider not only physicochemical characteristics of drugs but also delivery formulations and biological barriers in order to reach intracellular targets in the central nervous system (CNS) and to mitigate systemic side effects.
2. Organization of Mitochondria
Mitochondria consist of a double membrane with an intermembrane space and an internal mitochondrial matrix (MM) that contains the mitochondrial DNA (mtDNA). The outer mitochondrial membrane (OMM) contains the voltage-dependent anion channel (VDAC) and the permeability transition pore (mPTP) associated with the unspecific trans-location of small molecules (1-5 kDa) through passive diffusion [17][18][17,18]. The inner mito-chondrial membrane (IMM) is organized in folds or cristae and contains the electron transport chain (ETC) where the oxidative phosphorylation (OXPHOS) of adenosine di-phosphate (ADP) proceeds (Figure 1). The mtDNA, located in the MM, encodes the protein subunits of respiratory chain complexes I, III, IV, and V, along with RNA components for mitochondrial protein synthesis [19]. Complex II is encoded entirely by autosomal genes [20]. These ETC complexes catalyze redox reactions from reduced dinucleotide do-nors to molecular O2, generating a mitochondrial membrane potential (MMP) along the IMM by pumping protons from the MM to the intermembrane space. Finally, the return of protons into the MM through an ATP synthase present in IMM drives ATP synthesis from ADP and inorganic phosphate [21][22][21,22].
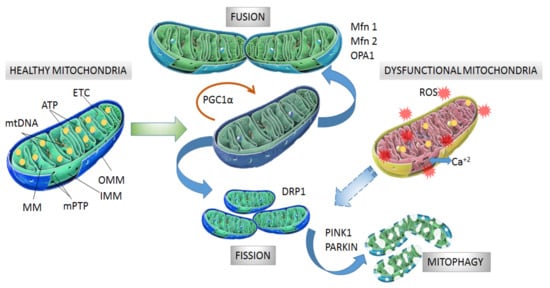
Figure 1. Structure and processes involved in dynamics of healthy and dysfunctional mitochondria. Healthy mitochondria display coordinated and dynamic processes of fusion and fission in order to regulate their morphology, size, and number. After mitochondrial biogenesis guided by PGC-1α protein, fusion generates an interconnected mitochondrial network, which is orchestrated by OPA1, Mfn1 and Mfn2 proteins. Fission results in small size mitochondria without mtDNA replication due to fragmentation and separation from the mitochondrial network, which is a process driven by dynamin- related protein (DRP1). Fragmented mitochondria are degraded by mitophagy, which is a process involving PINK1 and PARKIN proteins. Dysfunctional mitochondria showing alterations in structure and function in neurodegeneration are degraded by mitophagy. Mitochondrial dynamics are maintained by constant activity and precise balance between the biogenesis and clearance of fragmented and defective organelles. mtDNA: mitochondrial DNA, ATP: adenosine triphosphate, ETC: electron transport chain, MM: mitochondrial matrix, mPTP: permeability transition pore, OMM: outer mitochondrial membrane, IMM: inner mitochondrial membrane, PGC-1α: peroxisome proliferator activated receptor-gamma coactivator 1-alpha, Mfn1 and Mfn2: mitofusins 1 and 2, OPA1: optical atrophy 1 protein, DRP1: dynamin related protein, PINK1: PTEN-induced kinase 1, PARKIN: Parkin RBR E3 ubiquitin-protein ligase.
Since ETC operates under the presence of O2, the process involves one-electron redox reactions, which demonstrates that mitochondria are the major intracellular source of ROS under normal physiological conditions [23][24][25][26][23,24,25,26]. Indeed, mitochondria generate almost 90% of cellular ROS [3]. Under pathological conditions, altered ETC function causes an exacerbation of mitochondrial ROS production leading to bioenergetic impairment and cellular and tissue damage by oxidative stress; contributing to a progressive ND process [27][28][27,28].
A relevant aspect of mitochondrial biology is the exquisite regulation of calcium con-tent. Excessive levels of mitochondrial Ca+2 causes increased ROS production, mPTP open-ing and impairment of energetic function [1][29][30][1,29,30]. In addition, the disruption of mitochondrial contacts with membranes of the endoplasmic reticulum (ER) is a crucial event for mitochondrial integrity since function and structure are highly dependent on the flux of Ca+2 from and into the ER [31][32][31,32].
3. Mitochondrial Dynamics
Mitochondrial biogenesis is controlled by the transcriptional factor peroxisome proliferator activated receptor-gamma coactivator 1-alpha (PGC-1α) which is activated by di-rect interaction with NAD+-dependent deacetylase sirtuin 1 (SIRT1) [33] and phosphorylation by tAMP-activated protein kinase (AMPK) [34]. Then, phosphorylated PGC-1α is translocated into the nucleus where it promotes expression of the nuclear respiratory factors (NRFs) needed for further gene expression of mitochondrial proteins [35]. Among the mitochondrial proteins expressed are ETC protein subunits, fatty acid β-oxidation proteins and mitochondrial transcription factor A (mtTFA), which drive the transcription and replication of mtDNA. Additionally, NRFs also bind to promoter regions of genes coding for ROS scavengers [36][37][36,37].
The morphology, size and number of mitochondria are regulated by coordinated cycles of fusion and fission known as mitochondrial dynamics [38]. Mitochondrial fusion generates an interconnected mitochondrial network for the exchange of matrix contents and mtDNA molecules from healthy mitochondria donors to damaged mitochondria in order to reduce altered mtDNA. The main proteins with GTPase activity involved in mitochondrial fusion are optical atrophy 1 protein (OPA1), and mitofusins (Mfn1 and Mfn2) [39][40][39,40]. Mitochondrial fission generates smaller mitochondria without mtDNA replication by fragmentation and separation from the mitochondrial network followed by processing in the autophagosome [41]. Fission is modulated by GTPase dynamin related protein (DRP1), which is recruited from the cytosol to the OMM and interacts with fission 1 protein (Fis1) to stimulate mitochondrial fission [38][39][38,39] (Figure 1).
After mitochondrial fragmentation, the removal of altered mitochondria occurs by mitophagy, which is modulated by PTEN-induced kinase 1 (PINK1) and PARKIN RBR E3 ubiquitin-protein ligase (PARKIN) proteins [42][43][42,43]. PINK1 accumulates in the OMM in response to a reduction in MMP in dysfunctional mitochondria. Then, PARKIN is re-cruited from the cytosol to the OMM and promotes ubiquitination of mitochondrial proteins, leading to mitochondrial degradation [44][45][44,45]. Recently, it was observed that PINK1 accumulation and PARKIN recruitment are required to start mitophagy which is induced by slight oscillations in mitochondrial Ca+2 levels in human neuroblastoma SH-SY5Y cells [46]. In addition, PINK1 is also able to activate mitophagy directly without the participation of PARKIN by recruiting optineurin (OPTN) and nuclear dot protein 52 kDa (NDP52) [47]. In this case, PARKIN participates in further amplification of mitophagy in-duced by PINK1 [11] (Figure 1).
4. Mitochondrial Alterations Associated with NI&NDDs
Fragmented mitochondria with altered membrane structure have been found in the brain of patients with AD, which is a neurodegenerative disorder characterized by memory and learning impairment [48]. The dysregulation of mitochondrial Ca+2 content and accumulation of deformed mitochondria have been reported in animal models of HD [12][49][50][12,49,50], which is a dominant heritable pathology characterized by cognitive impairment, chorea, dystonia, and progressive loss of motor coordination [51]. mtDNA mutations affecting the ETC complexes have been reported in patients with MS [52], which is an autoimmune disease of the central nervous system (CNS) characterized by neuroinflammation, demyelination, and axonal damage [53]. In addition, several single nucleotide polymorphisms of mtDNA have been correlated with an increased risk of MS [54][55][54,55]. In comparison to healthy individuals, higher rates of mtDNA deletions were observed in the substantia nigra of autopsied brain samples from patients with PD [56], which is a neurodegenerative disease characterized by loss of dopaminergic neurons, leading to cognitive and motor alteration. Even though precise mechanisms determining how defective mitochondria promote the ND process are elusive, recent evidence has involved exacerbated mitochondrial ROS production in cellular toxicity and the promotion of aggregation and accumulation of toxic intracellular proteins. In turn, the accumulation of toxic proteins interferes with mitochondrial function, impairing energy production and maintaining an oxidative cellular unbalance that impacts the structure and function of the CNS [57][58][59][60][57,58,59,60]. In addition, the cytosolic release of mtDNA can induce the activation of the inflammatory response [61], leading to injury and functional impairment of the CNS. mtDNA from dysfunctional mitochondria also induces the activation of nucleotide-binding domain and leucine-rich repeat (NLR) pyrin domain containing 3 (NLRP3) inflammasome proteins, which is involved in cellular apoptosis [62]. Alternatively, the cytoplasmic release of proteins from dysfunctional mitochondria such as cytochrome c is able to promote cell loss in the CNS by apoptotic mechanisms [63][64][63,64]. Interestingly, a decreased anterograde and retrograde mitochondrial transport [65][66][65,66] has been involved in the subsequent structural alterations of axons and further morphological changes of mitochondria within the spinal cord of mice developing experimental autoimmune encephalomyelitis (EAE), an animal model of MS [65]. Thus, the impaired transport of mitochondria in neurons would limit the energetic supply needed to counteract demyelination and degenerative processes in the axonal terminal. Importantly, increasing mitochondrial transport from the neuronal cell body to the axon resulted in effective protection of demyelinated axons from degeneration [67].
4.1. Proteinopathies and Alteration of Mitochondrial Biology in NI&NDDs
Mitochondrial dysfunction associated with NI&NDDs is also facilitated by pathological accumulation of specific aberrant proteins as a result of nuclear gene mutations or abnormal protein processing leading to oligomeric and fibrillary aggregates. The anomalous accumulation of protein aggregates impacts mitochondrial structure and function either due to altered interaction with other subcellular organelles or dysregulation of processes involved in mitochondrial dynamics. The main protein aggregates related to proteinopathies are: amyloid β (Aβ) peptide and Tau protein in AD [68][69][70][71][68,69,70,71]; α-synuclein (α-syn) in PD [72][73][74][72,73,74], transactive response DNA-binding protein of 43 kDa (TDP-43) in AD and ALS [61][75][76][61,75,76]; Cu, Zn-superoxide dismutase (SOD1) in ALS [77][78][77,78]; and Huntingtin protein (Htt) in HD [50][79][80][50,79,80]. Table 1 summarizes the evidence relating these protein aggregates with mitochondrial dysfunction in ND diseases. α-Syn is a neuronal protein associated with the release of neurotransmitters and synaptic vesicles [81], and its misfolding and aggregation in structures referred to as Lewy bodies, particularly in dopaminergic neurons, is a hallmark of PD [81][82][81,82]. α-syn interaction with the mitochondrial structure is associated with an impairment of ETC activity, decreased MMP, mPTP opening and mitochondrial swelling as well as increased levels of mitochondrial ROS and neuron cell death [73][74][73,74]. In addition, α-syn interferes with the mitochondrial contacts with ER, leading to the disruption of Ca+2 flux and a reduction of ATP production [83]. Additionally, α-syn association with mitochondria results in downregulation of the predominant SIRT in the mitochondria (SIRT3) [72], which is a molecule that protects mitochondrial integrity and energetic function [84]. SIRT3 reduction is also accompanied by an increased expression of the fission protein DRP1 in neural cells and brain tissue of mice expressing α-syn [73]. α-Syn is also upregulated in the neurons and glia of demyelinating lesions in the spinal cord of mice developing EAE [85][86][85,86]. Moreover, the levels of α-syn in the cerebrospinal fluid of MS patients correlates with disease disability, suggesting a participation of α-syn in demyelinating and NI pathologies [87]. The intracellular accumulation of aggregates of Aβ peptide and hyperphosphorylated protein Tau are biochemical characteristics of AD [71][88][89][71,88,89]. The Aβ peptide derives from amyloid precursor protein (APP) in the mitochondria–ER contacts through sequential processing by BACE1 (β-site APP cleavage enzyme 1) and γ-secretase [90]. Aβ interacts with mitochondrial molecules and structures, which results in ROS production, mitochondrial dysfunction, and subsequent cell damage [91][92][93][94][91,92,93,94]. In addition, Aβ alters mitochondrial morphology and fragmentation by increasing DRP1 and reducing Mfn1 levels in cellular models [68][94][68,94]. Moreover, the mitochondrial fission promoted by Aβ occurs through O-GlcNAcylation of DRP1 in both neuronal cell lines and primary cultured neurons [69]. Interestingly, the activated form of DRP1 by O-GlcNAcylation is also found in the brains of mice in an AD mouse model [69]. Tau is a microtubule-binding protein in neurons, and its abnormal processing and aggregation by hyperphosphorylation promotes dissociation of preformed microtubules, interaction with mitochondrial ETC complexes, reduction of ATP production, and neuronal death [71][93][71,93]. In addition, hyperphosphorylated Tau interacts with VDAC1 mitochondrial protein promoting the alteration of energetic functions, Ca+2 homeostasis, and oxidative balance [71][91][71,91]. In addition, Tau alters mitochondrial fission and mitophagy by interacting with DRP1 and PARKIN proteins, respectively, in both patients and transgenic mouse models of AD [70][93][70,93]. TDP-43 is an essential ribonucleoprotein that can also form toxic cytosolic aggregates in AD. Diverse mutated forms of TPD-43 have been localized as aggregates in the mitochondria of mouse models and patients with familial ALS and are associated with structural and functional alterations in mitochondria [61][95][96][61,95,96]. Consistently, the inhibition of mitochondrial localization of TDP-43 restored mitochondrial function and ameliorated motor and cognitive deficits in ALS and AD models [75][76][97][75,76,97]. In HD, the generation of a mutated huntingtin protein (mHTT) by abnormal expansion of a CAG polyglutamine trinucleotide [98] impacted mitochondrial function and subsequent ND processes [50][79][80][50,79,80]. The enzyme SOD1 is mainly a cytosolic molecule, but mutated forms of SOD1 have been localized in aggregates associated with mitochondria in transgenic mouse models and patients with familial ALS [77][78][77,78]. Interestingly, the pharmacological reduction of misfolded SOD1 restored the structural integrity of mitochondria, reduced degeneration of motor neurons, and attenuated motor deficits in a transgenic ALS mouse model [99].
Table 1. Protein aggregates related to mitochondrial dysfunction in models of neurodegenerative diseases.
| Protein Aggregates |
Disease Model | Effect on Mitochondria | Refs | |||||
|---|---|---|---|---|---|---|---|---|
| α-synuclein | PD | ↑mitochondrial ROS levels, ↓ ETC activity, ↓stability of mitochondrial membranes, ↑mPTP opening, ↓mitochondria-ER contacts, ↑DRP1 and ↓mitochondrial SIRT3 levels (a protective molecule of mitochondrial integrity and energetic function [84]) | [72][73][74][83][100] | [72,73,74,83,100] | ||||
| Amyloid b | AD | ↑mitochondrial ROS levels, ↑mitochondrial fission (↓Mfn1, ↑DRP1 levels and ↑O-GlcNAcylation of DRP1) | [68][69][] | [68 | 91] | ,69 | [92][94 | ,91, 92,94] |
| Tau | AD | ↑microtubule dissociation, ↑mitochondrial ROS levels, ↓ATP production, ↑mitochondrial fission and ↓mitophagy (interaction with DRP1 and PARKIN proteins) |
[70][71][93] | [70,71,93] | ||||
| Transactive response DNA-binding protein of 43 kDa (TDP-43) |
AD, ALS | ↑mitochondrial ROS levels, ↓stability of mitochondrial structure, ↑mPTP opening |
[61][75][95][96][97] | [61,75,95,96,97] | ||||
| Huntingtin | HD | ↑mitochondrial ROS levels, ↓stability of mitochondrial structure, ↑mPTP opening, ↑mitochondrial fission by activation of DRP1, ↓mitophagy, ↑disruption Ca | +2 | flux between ER and mitochondria | [49][79[101] | [49 | ] [80] | ,79, 80,101] |
| Superoxide dismutase |
ALS | ↓mitophagy (by arresting optineurin protein), ↓stability of mitochondria structure, ↓flux of protein from and to the mitochondria | [102][103][104][105] | [102,103,104,105] |
ROS: reactive oxygen species, SIRT3: sirtuin 3, DRP1: dynamin related protein, α-syn: α-synuclein, ETC: electron transport chain, ER: endoplasmic reticulum, mPTP: mitochondrial permeability transition pore, Mfn1: mitofusin 1, PARKIN: PARKIN RBR E3 ubiquitin-protein ligase, PD: Parkinson’s disease, AD: Alzheimer’s disease, ALS: amyotrophic lateral sclerosis, HD: Huntington’s disease.
4.2. Alteration of Mitochondrial Dynamics in NI&NDDs
Mitochondrial dysfunction associated with NI&NDDs is also characterized by the altered activity of key proteins involved in mitochondrial dynamics. Diverse studies performed in cellular and animal models of NI&NDDs as well as in postmortem brain tissue of patients with NI&NDDs have shown increased activity of fission proteins such as DRP1 and FIS1 and in some cases reduced levels of Mfn and OPA proteins [50][69][80][94][101][106][50,69,80,94,101,106]. Interestingly, reversing DRP1 activation by pharmacological or genetic inhibition reduced mitochondrial fission and cell death in cellular and animal models of ND diseases [107][108][109][107,108,109]. Alternatively, increasing the mitochondrial fusion through overexpression of Mfn2 restored mitochondrial dynamics and attenuated neural damage and motor deficits in NI&NDDs [110].
In addition, impaired mitophagy has been observed in several studies with animal models of NI&NDDs [46][79][111][46,79,111]. Dysfunctional mitophagy has been associated with altered aspects of PINK and PARKIN function; such as their inactivation, deficient expression, suppression of mitochondria calcium signaling hampering recruitment of PINK and PARKIN to the mitochondria [46], or gene mutations affecting these proteins. Mutations in PINK and PARKIN genes have been related to the origin of familial PD, which can represent around 10% of the diagnosed forms of PD [112][113][114][112,113,114]. Interestingly, normalizing PINK and PARKIN activity through chemical agents restored mitophagy and cognitive impairment in ND models [13][115][13,115]. Additionally, PINK1 overexpression rescued mitochondrial dysfunction and restored the removal of defective mitochondria in transgenic animal models of AD and HD [11][12][116][11,12,116].
Recently, a rat model of PD induced by direct intracerebroventricular injection of 6-hydroxydopamine (6-OHDA) exhibited accumulated defective mitochondria as spheroids in dopaminergic neurons by unfinished mitophagy. Interestingly, altered mitochondria were transferred to surrounding astrocytes in order to complete the remaining steps of mitophagy [117]. The cellular expulsion of damaged mitochondria has also been observed in a PD model of neuronal cells treated with rotenone and in fibroblasts and cerebrospinal fluid samples from PD patients carrying a PARKIN mutation [118]. A self-destructive process of mitochondrial removal referred to as mitoautophagy has been proposed to operate in the motor neurons of an ALS transgenic mouse model. Unlike classical mitophagy, mitoautophagy produces mitochondrial degradation without the participation of lysosomes or autophagasomes [105].
Decreased mitochondrial biogenesis is observed in both patients and animal models of ND diseases [119][120][119,120]. The impaired process is characterized by the altered expression of PGC-1α, which is the main molecule involved in mitochondrial biogenesis. PGC-1α expression is significantly reduced in the brain tissue of HD mice [80] as well as in postmortem brain tissue of AD [121] and MS patients [122]. Additionally, PGC-1α has been found post-translationally inactivated by phosphorylation in the neurons of spinal cords from EAE mice [14]. Consistently, mice overexpressing neuronal PGC-1α showed an increased number of active mitochondria with enhanced respiratory capacity and a significantly better recovery of clinical disability and neurodegeneration induced by EAE compared to wild-type controls [14].
4.3. Energy Impairment Associated with Mitochondrial Dysfunction in NI&NDDs
A common hallmark of different NI&NDDs is the bioenergetic deficit due to mitochondrial dysfunction. ETC activity and ATP production declines in mitochondria in early stages of AD and ALS [123][124][123,124] and an altered brain energy metabolism, evidenced by a reduced neuronal glucose uptake, is associated with the progression of neurodegeneration and frequently manifests before symptomatic onset in AD, PD, ALS, and HD [125][126][127][128][129][125,126,127,128,129]. In addition, brain glucose hypometabolism correlates with subsequent patterns of motor and cognitive deficits along with pathology progression [125][130][131][125,130,131]. Reduced glucose uptake in the brain of patients with ND diseases associates with brain insulin receptor desensitization, as evidenced by the significant reduction and deterioration of insulin receptors in AD [132][133][132,133]. Interestingly, insulin regulates the function of mitochondria by upregulating the ETC complex proteins [134] and brain insulin resistance has been related to mitochondrial dysfunction and promotion of PD [135]. Consistently, evidence (see Section 6) shows that compounds that promote uptake of glucose as well as insulin sensitizers restore mitochondrial function and ameliorate cognitive and motor disturbances in ND mouse models. Therefore, an energy impairment promoted by the hypometabolism of glucose and dysfunctional mitochondria contributes to ND diseases [136][137][138][136,137,138].
4.4. Oxidative Stress Associated with Mitochondrial Dysfunction in NI&NDDs
Impaired operation of ETC is observed in the mitochondria of neurodegenerative tissues leading to increased ROS production and oxidative stress. The brain is very sensitive to redox impairment due to the high content of polyunsaturated fatty acids. Therefore, the brain is prone to suffer further oxidation events due to the presence of transition metals, such as iron and copper, putting mitochondria in a circular loop of damage and leading to more defective mitochondria [9][10][9,10]. Thus, dysfunctional mitochondria can exacerbate the oxidative environment in ND diseases. In turn, oxidative conditions severely affect the machinery involved in energy production exacerbating ROS production in mitochondria. For example, the expression of mtDNA genes required for OXPHOS and ETC function are significantly reduced in postmortem brain tissue of AD and PD [121] as well as in MS patients and in motor neurons of the spinal cords of EAE mice [52][57][139][140][52,57,139,140]. In these cases, reduced mitochondrial function is accompanied by an augmented oxidative response which often precedes more severe signs of biochemical and clinical defects in memory and neurodegeneration [57][58][57,58]. Studies have determined reduced activity of the complex II enzyme succinate dehydrogenase (SDH) and a decreased complex II-III activity in the basal ganglia of HD patients along with a decrease in complex IV activity in HD striatum. 3-nitropropionic acid (NPA), an inhibitor of SDH, causes mitochondrial damage leading to an increase in electron leakage from the mitochondria, production of ROS and nitrogen species, and depletion of antioxidant defenses. Interestingly, 3-NPA in rodents causes striatal degeneration and impairment in locomotor activity, body weight, and cognitive deficits that closely mimic symptoms seen in HD [141][142][141,142]. PD induced in mice or primates by diverse compounds such as 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP), rotenone or 6-OHDA produces loss of dopaminergic neurons by inhibition of ETC in the mitochondria and exacerbated generation of ROS [143][144][145][146][143,144,145,146]. Impaired mitochondrial production of ATP in motor neurons in mouse models and patients with ALS is accompanied by massive oxidative damage prior to the manifestation of clinical symptoms or at early stages of disease [60][147][148][60,147,148].The oxidative stress can also induce mitochondrial dysfunction by affecting mitochondrial dynamics. Reactive nitrogen species can activate DRP1, leading to mitochondrial fragmentation and ND damage in cellular and animal models of AD and MS [148][149][148,149]. Interestingly, treatment with chemical antioxidants reverted the defective operation of mitochondrial OXPHOS in cultured fibroblasts from ALS patients [16]. Thus, counteracting the oxidative stress associated with mitochondria dysfunction could be an important therapeutic strategy for tackling ND processes.