Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Telma de Sousa and Version 2 by Rita Xu.
Pseudomonas aeruginosa has the ability to infect not only humans, but also animals and plants. Given its role as an important human pathogen, several virulence factors have been studied, as well as their regulatory systems.
- Pseudomonas aeruginosa
- pathogenicity
- multidrug-resistant
1. Introduction
One of the current greatest global public health problems is the pathogenesis associated with the emergence of generalized infectious bacteria [1]. This pathogen is armed with a wide set of virulence factors, allowing it to harm host cells and modulate human adaptive immune mechanisms, thus causing the appearance of new infections [2][3]. The T3SS system is one of its main virulence factors, and a very complex matrix of environmental signals is integrated to tightly control the injection of proteins into host cells, where they are directed to specific compartments and manipulate a number of cell processes [3][4]. However, the pathogenesis of this bacterium is not exclusively focused on the emergence of new infectious diseases, but also on biofilm formation and on the appearance of new antibiotic-resistant strains, or on the increase in prevalence of already known strains [4][5]. The acquisition of resistance to two or more classes of antibiotics by pathogenic bacteria is denominated as multidrug-resistance (MDR) [5][6]. P. aeruginosa is one of the bacterial species that have developed an alarming number of multidrug-resistant strains. These strains are associated with significant morbidity and mortality and they are responsible for 10% of nosocomial infections [6][7].
2. Pseudomonas aeruginosa
Pseudomonas aeruginosa is a ubiquitous bacterium, present both in terrestrial and aquatic ecosystems. As a result, it can be found in many foodstuffs but also in hospital environments. P. aeruginosa is considered to be a bacterium of major medical importance due to its great ability to adapt to different environments, but also because of its ability to cause chronic infection in vulnerable people [7][8]. The first report describing the genus Pseudomonas came from the German botanist Walther Migula in the late 19th century. He visualized mobile cells with spores, such as the nanoflagellate Monas spp. Thus, the name Pseudomonas was generated from the identification of this false or “pseudo” Monas spp. nanoflagellate [8][9]. However, the specific name “aeruginosa” was described earlier in 1872 by Schroeter because of the color of the colonies on certain media, resembling copper rust or verdigris color, hence being green. Schroeter attached the name to the genus Bacterium, to give Bacterium aeruginosum, and later, Migula transferred the species to the newly described genus Pseudomonas [9][10].
P. aeruginosa is a gram-negative, bacillus-shaped bacterium (size from 0.5 to 3.0 µm), with an aerobic metabolism and a single flagellum that helps in move. It is a non-fermentative bacterium which, in aerobic situations, uses the glycolytic pathway for glucose degradation, with oxygen as its final electron acceptor. However, under anaerobic conditions, nitrogen can be used as an electron acceptor. Furthermore, ATP acquisition is achieved by the action of several membrane ATPases reusing ADP and H+ from previous reactions. It should be noted that sometimes, aromatic compounds like phenolsulfates and phenylalanine can be considered as putative substrates for this species’ catabolic activity [10][11]. This species can survive in a wide range of environmental conditions (mostly regarding their chemical composition), it can use a variety of different carbon, nitrogen and energy sources, but it has a rather limited range of growth temperatures [9][10].
2.1. Bacterial Pathogenesis
P. aeruginosa is adaptable to different environments and metabolically versatile, covering a wide variety of habitats, including the human body, soil, water, hospital environment, drains, but also other water-rich locations (swimming pools), as well as non-household environments (such as river water), and can also be isolated from vegetables [11][12][13][14][12,13,14,15]. This is due to the minimum nutritional requirements necessary for the growth of this bacterium and its ability to survive under a variety of environmental and physical conditions [10][13][11,14]. The dissemination of new infections that come from the environment and hospitals requires special attention in order to minimize the chance of nosocomial infection by this pathogen [12][15][13,16].
The ubiquity of P. aeruginosa in nature comes from its natural ability to use several mechanisms to resist against other organisms. For example, when using the type III secretion system to directly inject cytotoxic effector proteins into the cytosol of eukaryotic cells, a polysaccharide-encased community (a biofilm) of P. aeruginosa can be formed and this community can resist to predation by protozoa [16][17][17,18].
The infection potential of P. aeruginosa comes in part from the presence of virulence factors and the ability to metabolize many antibiotics (encoded in genes that are collectively organized in genomic islands), consequently, promoting infectivity potential [18][19][20][19,20,21]. In general, virulence factors can be classified as cell- associated or extracellular virulence factors [21][22][22,23], both being secreted via secretion systems. Cell-associated virulence factors are important for the bacterial colonization process and they are secreted and bound to the cell envelope (inner or outer membrane) [17][18]. On the other hand, extracellularly virulence factors are released by P. aeruginosa after colonization and they are secreted into the extracellular medium (exoproteins) [17][18].
2.2. Clinical Impact of Pseudomonas aeruginosa
P. aeruginosa is an opportunistic pathogen. It lives as a commensal and environmental organism, but can occasionally change to a pathogenic state, causing infections that are difficult to treat [23][24]. For example, P. aeruginosa is the major cause of infections such as ventilator-associated pneumonia, urinary tract, bloodstream, and chronic infections [24][25][26][25,26,27]. In immunocompromised hosts, like AIDS or diabetes mellitus patients, bacteremia caused by this bacterium is a common complication [25][26]. In animals, P. aeruginosa causes diseases in both livestock and pets. For example, it causes otitis and urinary tract infections in dogs, mastitis in dairy cows, hemorrhagic pneumoniae in fur-bearing animals such as mink or foxes, and endometritis in horses [27][28].
This bacterium displays a natural resistance to many classes of antibiotics and its capacity to rapidly develop new ones during treatment represents one of the most common reasons for therapeutic failures [28][29]. P. aeruginosa is one of the six “ESKAPE” pathogens belonging to the World Health Organization “priority pathogens” list of antibiotic resistance [1]. The resistance rate of P. aeruginosa to antibiotics is increasing worldwide and that is why it is necessary to increase monitorization of infections in animals and humans in order to minimize possible transfers between hosts [27][28].
P. aeruginosa can also develop and grow in a sessile community structure that provides protection against antibiotics, host defense mechanisms, desiccation, ultraviolet light, and disinfectants [29][30]. Given its resistance to antibiotics, the opportunistic, and resilient character, it has become one of the most concerning issues of pathogenesis nowadays and has drawn the attention of many researchers in the quest to obtain more knowledge and understanding [6][30][31][7,31,32].
3. Genome Structure in Pseudomonas aeruginosa
The genetic repertoire of P. aeruginosa reflects the adaptative character of this bacterial species [10][11]. The metabolic versatility is provided by genes encoding enzymes that participate in the metabolic pathways, transcriptional regulators, and regulatory systems [32][33]. The whole genome sequence of P. aeruginosa strain type PAO1 was published by Stover and other collaborators in 2000. Thereafter, several strains have had their whole genomes sequenced [33][34]. The P. aeruginosa PAO1 genome is used as a reference for comparing with the genomes of other strains [33][34]. P. aeruginosa PAO1 has a very large and complex genome, around 6.3 Mbp (G + C content 66.6%), encoding 5700 genes, including 5584 predicted open read frames (ORFs) [33][34][34,35]. In addition, it is estimated that 150 of the genes identified in P. aeruginosa PAO1 encode outer membrane proteins related to adhesion, movement, antibiotics, and virulence factor output (Figure 1) which represents a much higher number when compared to other genomes [35][36].
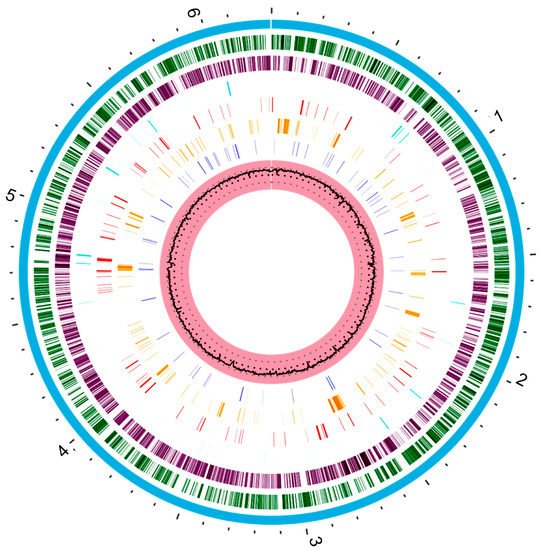
Figure 1. The circular representation of the P. aeruginosa PAO1 genome generated with www.patricbrc.org (accessed in 8 May 2021). This genome contains about 6.3 Mbp. The pink circle that contains a black circle in the middle is the G + C content percentage (66.6%). The dark blue bars represent a drug target. Orange bars represent genes encoding virulence factors and the red bars genes encoding antimicrobial resistance. Cyan bars represent non-coding regions. The purple and dark green circle represent the reverse and forward coding regions of the genome, respectively. Finally, the blue circle that covers all regions represents the entire chromosome.
P. aeruginosa has a mosaic genome, composed of many core genes interspersed by strain-specific blocks of genes [36][37][37,38]. As a result of comparative genomic studies within the species, the genome of P. aeruginosa has been classified in three groups depending on its characteristics: the core genome, the accessory genome, and the pan-genome [17][34][36][37][38][18,35,37,38,39].
The core genome defines the region of the genome possessed by almost all P. aeruginosa strains, interspersed with “accessory” genomic elements that are present in some, but absent in other strains of the same species [32][34][33,35]. The core genome regions represent approximately 90% of the total P. aeruginosa genome, display low levels of genetic diversity (0.5–0.7%), contain the majority of genes with housekeeping functions, and do not tolerate excessive changes over short evolutionary periods [39][40][41][40,41,42].
The accessory genome of P. aeruginosa consists of non-conserved, variable-length stretches of DNA, generally located in extrachromosomal elements, and blocks of inserted DNA in certain loci [32][42][33,43]. These DNA segments tend to cluster at certain loci, rather than being randomly distributed throughout the core genome [39][40]. These loci are often referred as genomic islands (>10 kb) [42][43]. This part of genome is very relevant for its clinical implications, since it harbors genes that encode proteins with homologies to virulence factors and genes that encode resistance to various classes of antibiotics [38][43][39,44]. For example, genomic island of P. aeruginosa PAPI-2 contain the gene encoding ExoU, a type-III secreted effector protein linked to increased virulence in animal models and human patients [34][35]. The intrinsic determinants of antibiotic resistance in P. aeruginosa, such as efflux pumps and a β-lactamases, are located in the core genome. However, genes of acquired antibiotic resistance are present in the accessory genome [17][30][18,31]. Indeed, the dissemination of multidrug-resistant strains is often due to the resistance genes present in the accessory genome [38][39]. Therefore, identification of these accessory genomic elements in P. aeruginosa has a major impact on the study of the evolution, adaptation, and infectious potential of this organism [34][35].
The general architecture of the P. aeruginosa pan-genome can be represented as a circular chromosome with polymorphic strain-specific segments, flanked by conserved genes referred to as anchors [39][40]. Basically, this genome consists of genes within the core genome and accessory genome, with the latter containing all “dispensable”, strain-specific genes present only in a subset of strains [44][45]. The size of the pan-genome currently exceeds 10 Mbp, whereas the core genome is roughly 5.84 Mbp, representing 89.7% of the total genome, and the accessory genome is about 727 kbp, representing, in average, 11.1% of the total genome [34][35]. The P. aeruginosa pan-genome contains approximately 54,272 genes, 665 of which are core genes, 26,420 are accessory genes, and 27,187 are unique genes (present in one strain only) [38][39].