Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Maria Vulf and Version 3 by Lindsay Dong.
Subclinical inflammation in morbid obesity is associated with the activation of the immune system and the development of concomitant diseases. Impaired immune homeostasis and dysregulation of immune cells in adipose tissue are associated with phenotypic and functional changes in the pool of T-lymphocytes and the development of chronic vitamin D deficiency. Low vitamin D levels in obesity lead to activation, proliferation, and production of pro-inflammatory mediators T cells. Hypovitaminosis D is the reason for the decrease in the functional potential of regulatory and anti-inflammatory lymphocytes and the maintenance of the inflammatory response.
- vitamin D
- autoimmune disease
- Th17 cells
- inflammation
- obesity
1. Introduction
The pathogenesis of obesity is closely related to changes in the homeostasis of immune cells in the intestine, adipose tissue, and liver [1][ 1 ]. A high-calorie diet causes the development of pathogenic strains of microorganisms in the gastrointestinal tract and a violation of the density of endothelial cells with the development of metabolic endotoxemia [2][3][ 2, 3 ]. Microbial dysbiosis leads to the dysfunction of immune cells and increases the body's susceptibility to infections [ 4 [4]].
Obesity is associated with chronic sluggish inflammation with localized distinct foci of inflammation in the visceral adipose tissue and liver [ 1 [1]]. T cells are an important pathogenetic component of inflammatory diseases [ 5 [5]]. It was found that obesity and type 2 diabetes mellitus are associated with a significant increase in Th1 and Th17 lymphocytes in visceral and subcutaneous adipose tissue (and a change in the cellular composition depending on the location of adipose tissue) against the background of a decrease in the number and functional properties of T-regulatory (Treg) cells [ [6]6 ].
It should be noted that hyperplasia and hypertrophy of adipose tissue activate stress signals in adipocytes, which contribute to an increase in the secretion of free fatty acids and reactive oxygen species (ROS) [ [7]7 ]. Oxidative stress associated with obesity and autoimmune diseases (AID) activates the mTORC1 pathway, which provides glucose uptake and aerobic glycolysis by modulating the transcription factor HIF-1α [ [8]8 ]. The mTORC1-HIF-1α pathway stabilizes and enhances the transcriptional activity of the main regulator of the development and differentiation of Th17 cells, RORyt, under hypoxic conditions [ [9]9 ].
Obesity is associated with increased death of adipose tissue cells, which leads to an increase in extracellular ROS, which acts as an alarm (danger) [10][ 10 ]. It has been shown that ROS regulate Th17 cell responses via the P2X7 purinergic receptor [ 11, [11][12]12 ]. Increased expression of the P2X7 receptor in visceral and subcutaneous adipose tissue in people with metabolic syndrome confirms the role of adipose tissue in Th17 lymphocyte differentiation [ 12 [12]]. КIn addition, it is likely that low serum vitamin D levels are associated with obesity and related diseases [ [13]13 ].
Vitamin D has been found to have a broad spectrum of functions. One of these is to act directly on immune system cells by regulating their proliferation and metabolism [14][17]. Activated T lymphocytes express the nuclear and cytoplasmic vitamin D receptor (VDR) [15][14]. Cytoplasmic ligation of VDR and transfer of the formed complex into the nucleus contribute to the expression of pro-inflammatory genes (sensitive to the action of vitamin D, namely the response elements of the vitamin D receptor regions (VDRE), CTLA-4, PLC-y1, IL-13, IFNy, IL-17A, IL-17F, IL-26) in T lymphocytes [13][16][13,18].
Interestingly, hypovitaminosis D induces the differentiation and development of Th17 cells [14][17][17,19]. Moreover, constant antigenic stimulation and a pro-inflammatory microenvironment against the background of chronic inflammation contribute to an accelerated formation of a memory T cell pool, which plays an important role in the pathogenesis of many serious diseases. Memory T cells formed in the long-term inflammatory process (e.g., in obesity) may acquire new functional properties.
Considering that Th17 lymphocytes may constitute a significant part of the memory T cell pool, the molecular processes involving vitamin D (hypovitaminosis), which affect their development, could serve as a fundamental basis for understanding the pathogenesis of non-infectious inflammatory conditions and for finding specific targets to prevent the formation of T cells with autoreactive properties. In addition, Th17/T memory cells may be a link between the state of vitamin D deficiency and the development of obesity, as well as biomarkers for assessing (the degree of) the risk of developing more severe complications.
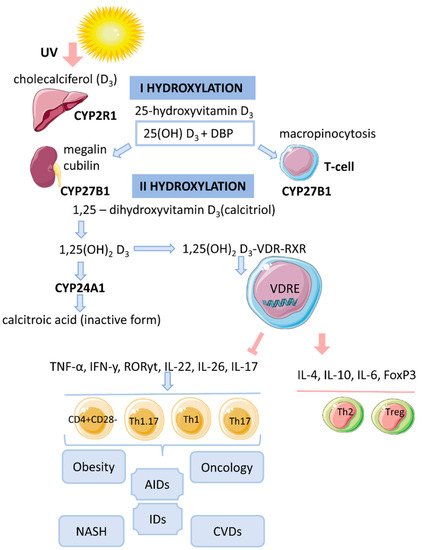
2. Vitamin D Metabolism
Vitamin D is a fat-soluble secosteroid, 70–80% of which is synthesized in a photolysis reaction in the skin [18][20]. Under the influence of ultraviolet radiation (UV) from the B spectrum of the sun, the process of photosynthesis in the epidermis is activated. In this process, 7-dehydrocholesterol (provitamin D) is converted into previtamin D3, which undergoes isomerization of three double bonds after a thermochemical reaction, forming the inactive vitamin D3 (cholecalciferol). The synthesis of vitamin D3 takes up to 3 days from the time the skin is exposed to UV rays. This process is the main source of vitamin D and it depends on the intensity of UV radiation (latitude, season, time of day), skin pigmentation and air pollution [18][19][20][20,21,22] (Figure 1).Figure 1. The mechanism of action of vitamin D on T cells in obesity.
3. Cytosolic Complex 1,25(OH)2D3-VDR-RXRF
The biological effect of 1,25(OH)2D3 is related to the activation of the cytosolic VDR receptor, whose active ligand is DBP and an indirect change in the transcriptional activity of vitamin D-associated genes [14][17]. VDR is a transcription factor and belongs to the family of nuclear receptors for steroid hormones (receptors for retinoic acid, thyroid hormones, sex hormones, adrenal hormones). The human VDR gene consists of eight coding exons (two non-coding) and two promoters and is located on chromosome 12 [19][21]. The VDR protein contains 427 amino acid residues. It acts as an obligate heterodimer that interacts with the retinoid X receptor (RXR), which subsequently causes the translocation of the complex into the nucleus and binds in a ligand-dependent manner to the promoter regions of target genes that are sensitive to vitamin D [14][18][17,20]. The functional domains of VDRs are the highly conserved NH2-terminal DNA-binding domain (DBD) and the more variable COOH-terminal ligand-binding domain (LBD). The DBD is a region with two zinc fingers, each containing a zinc atom in a tetrahedral arrangement with four invariant cysteine residues [19][21][22][21,30,31]. The lipophilic molecule 1,25(OH)2D3 can pass through the cell membrane and interact with the VDR [18][20]. Binding of 1,25(OH)2D3 to the VDR leads to conformational changes in the structure of the VDR protein that facilitate interaction with RXR and coregulatory complexes involved in the transcription of target genes [19][21]. Subsequently, the cytosolic complex 1,25(OH)2D3-VDR-RXR migrates to the nucleus, where it interacts with VDRE [15][14] (Figure 1). In the nucleus, the 1,25(OH)2D3-VDR-RXR complex interacts with histone acetyltransferases, transcriptional coactivators, corepressors and chromatin-restructuring complexes to modulate the transcription of target genes [16][18].4. Genomic Mechanism of Action of Vitamin D (T-lymphocytes)
After the identification of VDR in activated T lymphocytes, vitamin D has been proposed as a regulator of the immune system [13]. The active form of vitamin D, 25(OH)2D3, has an immunomodulatory effect on many components of the innate and adaptive immune systems [14][17]. 1,25(OH)2D3 regulates differentiation and maturation of subpopulations of innate immunity cells, antigen presentation and the production cytokines and chemokines. 1,25(OH)2D3 inhibits the inflammatory response by suppressing the expression of Toll-like receptors 2 and 4 (TLR2/4) and the secretion of pro-inflammatory mediators (IL-1, IL-6, TNF-α). In addition, the active form of vitamin D negatively regulates the differentiation, maturation and immunomodulatory capacity of dendritic cells (DCs) by reducing the expression of MHCII, CD40, CD80, CD86 and the maturation proteins CD1a, CD83 [14][17]. 1,25(OH)2D3 also inhibits the DC-mediated activation process of T cells and decreases the expression of inflammatory mediators (IL-12, IFNy in DCs) [5][18][5,20]. 1,25(OH)2D3 inhibits the proliferation of T lymphocytes and the production of IFNy and IL-17 and increases the secretion of IL-4 and IL-10. Thus, 1,25(OH)2D3 enhances the regulatory Th2 immune response and induces the differentiation of Treg cells, thereby reducing the pro-inflammatory potential of Th1 and Th17 cells. The complex 1,25(OH)2D3-VDR-RXR blocks the formation of NFAT/AP-1. It is known that the formation of the NFAT/AP-1 complex is necessary for the activation of the IL-2 promoter. The repressive effect of 1,25(OH)2D3 on IFNy gene transcription is due to the direct interaction of VDR-RXR with the silencer regions on the gene promoter. 1,25(OH)2D3 enhances the production of IL-4 by Th2 cells and potentiates the regulatory properties of Treg cells by activating the expression of the transcription factor FoxP3 [5][20][5,22]. The immunomodulatory effect of 1,25(OH)2D3 is also associated with an increase in IL-6 secretion, which may lead to a shift in the balance towards the Treg cell response [18][23][20,32]. 1,25(OH)2D3 prevents IL-17 production by suppressing inflammation and Th17-mediated autoimmunity [24][25][33,34]. The mechanism of IL-17 suppression by 1,25(OH)2D3 is based on blocking NFAT binding to the IL-17 gene promoter, sequestration of Runx1 factor by the VDR, recruitment of histone deacetylase (HDAC) and induction of FoxP3 expression [20][22]. In addition, the VDR interacts with the P105/P50, P100/P52 and P65 proteins of the NF-kB factor.In hypovitaminosis D, the addition of 1,25(OH)2D3 has been shown to help suppress the formation of inflammatory infiltrates and inhibit the expression of RORyt/IL-17 in spleen tissue of mice by preventing the translocation of P65 into the nucleus [26][35].
The stimulatory effect of 1,25(OH)2D3 on the production of anti-inflammatory cytokines (IL-4, IL-10) may be indirect and dependent on numerous intercellular contacts and the state of cell activation.
DCs are known to be an important target for 1,25(OH)2D3. Under the influence of vitamin D, their maturation is affected by transcription-mediated reprogramming of metabolic pathways (simultaneous increase in glycolysis and oxidative phosphorylation). Moreover, the mechanism for reducing IL-12 production in DCs involves binding of VDR-RXR to the NF-kB site on the IL-12p40 gene promoter. Thus, 1,25(OH)2D3 can alter T cell behavior by regulating DCs, causing T lymphocyte anergy [18][20] and converting pro-inflammatory Th1/Th17 cells into more tolerant cells (Th2/Treg cells) [19][21].
5. Vitamin D and T-lymphocytes
Expression of VDR has been detected in cells of innate and adaptive immunity [18][20]. Vitamin D exerts an immunoregulatory function by inhibiting proinflammatory cells and promoting the development of anti-inflammatory populations, thus participating in the processes of immune tolerance. Cells of the immune system express the enzyme CYP27B1 and can locally convert 25(OH)D3 to the active form 1,25(OH)2D3, where it is further utilized or locally released to neighboring cells [13][15][13,14]. However, the importance of the systemic circulating level of 25(OH)D3, which has a longer half-life, has been demonstrated [15][14].
The degree of activation of T lymphocytes is directly related to their phenotypic characteristics and functional potential. During their life (maturation), T cells (CD4+) undergo several stages of differentiation, starting with naïve (Tn) and then sequentially progressing into the group of effector cells (Teff) (including Th1, Th2, Th9, Th17, Th22 cells), central memory cells (Tcm)/memory effector cells (Tem) and terminally differentiated memory T cells (Temra) [27][36]. The level of VDR expression was found to be related to the activation of T lymphocyte; naive T cells are thus insensitive/resistant to vitamin D.
5.1. Th1 and Th2 Cells
Studies on obesity and DM2 have shown a significant increase in Th1 and Th17 cells and a decrease in the number and functional properties of Treg cells [6].
1,25(OH)2D3 was found to suppress IFNy production [16][18], blocking Th1 cell differentiation and development, against a background of increased secretion of IL-4 (which stimulates Th2 cell formation) [13].
5.2. Non-Pathogenic Th17 Cells and Treg
Obesity caused by a high-fat diet results in low secretion of Th17-associated cytokines in the intestine and adipose tissue, but high in the liver [28][29][37,38].
Non-pathogenic Th17 lymphocytes have a high intracellular content of polyunsaturated fatty acids and cholesterol esters and also a low content of saturated and monounsaturated fatty acids and triglycerides [30][39]. Melatonin, cholesterol, cholesterol sulfate and steroid lipids (oxysterols) have been shown to be ligands of RORα (RORyt) [31][40].
A distinctive feature of Th17 cells (compared to Treg cells) is the de novo synthesis of neutral fatty acids. The energy cost of this process in Th17 cells is compensated by glycolysis [32][41].
1,25(OH)2D3 has been shown to inhibit Th17 cell differentiation and stimulate Treg cell development [13]. 1,25(OH)2D3 inhibits the secretion of IL-17A, IL-17F, IL-22 and IL-26 [16][18].
5.3. Th1.17-Cells
Consumption of high-calorie foods leads to the disruption of immune cell homeostasis in the gastrointestinal tract and the formation of a pro-inflammatory pool of Th17 lymphocytes that can migrate to adipose tissue and liver. The development of an inflammatory response against a background of metabolic endotoxemia and adipocyte hypertrophy/hyperplasia can activate the plastic properties of Th17 cells, which, under similar conditions, can first transform into IL-17+IFNy+ lymphocytes and then into pathogenic Th17 cells (pTh17/Th1.17/nonclassical Th1) involved in the development of AIDs [33][42].
Th1.17 cells have been found to form a heterogeneous population that differs in phenotypic characteristics and cytokine-producing properties and depends on epigenetic stimuli that regulate permissive (H3K4me3) and repressive (H3K27me3) histone marks [23][34][32,43]. One of the characteristics of pTh17 lymphocytes is a high production of pro-inflammatory mediators: IFNy, TNF-α, GM-CSF and cytotoxic molecules (granzymes, perforin), together with a low secretion of IL-17, which is also reflected in the genotypic characteristics of these cells [35][44].
It was found that 1,25(OH)2D3 suppresses DC maturation and reduces the number of IL-17+IFNy+ cells during in vitro cultivation [36][45].
5.4. Memory T Cells
Vitamin D is an important regulator of CD4+ lymphocyte differentiation [13]. A link between vitamin D deficiency and an increase in the number of proinflammatory memory CD4+CD28 cells has been suggested [37][16]. In NASH, often associated with obesity, an increase in local infiltration of CD4+CD28 memory cells into the liver has been observed [38][46].
Loss of the co-stimulatory molecule CD27 (as well as CD28) by T cells (transition to the memory T cell pool) is associated with increased gene expression (mTORC1, ICC, cholesterol metabolism and glycolysis) [39][47].
This allows us to consider the population of Th1.17 cells that have undergone multiple antigenic stimulation as part of the memory T cell pool.
CD28null cells are newly activated memory/effector cells equipped with combinations of adhesion molecules and chemokine receptors that mediate the invasion of liver tissue. Upon reactivation, CD4+CD28null cells secrete high levels of proinflammatory cytokines TNF-a and IFNy [38][46]. Furthermore, pathogenic memory T cells express a multidrug resistance receptor (MDR-1 or P-glycoprotein) on the cell membrane, similar to cancer stem cells that are resistant to chemotherapy. 1,25(OH)2D3 decreases the drug resistance of tumor cells [40][51]. Vitamin D may have a similar effect on pathogenic CD4+CD28null cells in inflammatory diseases and AIDs.
Vitamin D has been found to decrease TNF-a production and to have a direct anti-inflammatory effect on CD4+CD28null cells that accumulate in the liver in primary sclerosing cholangitis [38][46].
An important finding is the extent of involvement of memory T cells, including Th1.17 cells, in peripheral blood and inflammatory foci in AIDs. Circulating memory T cells are less committed and actively respond to the anti-inflammatory effects of 1,25(OH)2D3 and block IL-17 and IFNy. However, memory T cells localized in areas of active inflammation are the most highly committed Th1,17 cells (probably Temra) that are resistant to the action of 1,25(OH)2D3 [5].
Thus, the final affiliation of memory T cells to a particular phenotype plays a central role in attenuating the anti-inflammatory effects of vitamin D. VDR, as a transcription factor, regulates the expression of genes (IL-17,IFNy) containing functional vitamin D response elements (VDRE) [41][42][52,53]. This process is regulated by genomic variations and epigenetic mechanisms that lead to specific changes in 1,25(OH)2D3-mediated chromatin remodeling and VDRE availability [43][54].
6. Hypovitaminosis D in Obesity
Based on previously obtained data, theories have been formulated that partially explain the relationship between hypovitaminosis D and obesity [44][45][46][55,56,57].- (1)
-
Volume dilution. Serum vitamin D levels decrease with increasing body size [47][48][58,59].
- (2)
-
Sequestration of vitamin D in adipose tissue. Vitamin D (synthesized by skin, supplements/medications) becomes tightly bound in fat stores and does not enter the bloodstream in sufficient quantity to maintain serum levels of 25(OH)D3 [44][46][55,57].
- (3)
-
Different ability to activate vitamin D in adipose tissue of lean and obese individuals [47][58]. In adipocytes, high expression of the enzymes 1α-hydroxylase (mitochondrial CYP27B1) and 25-hydroxylase CYP2J2 was found [44][55]. However, their expression is lower in obese people compared to lean people [49][60].
7. Molecular Mechanism of Action of Vitamin D on T Cells under Hypoxic Conditions Associated with Obesity
In obesity, the formation of local foci of hypoxia is observed. At the same time, hypoxia activates the transcription of the factor HIF-1α, which, together with mTORC1, promotes pTh17 cell differentiation [32][41]. Vitamin D has been shown to suppress LPS-induced expression of HIF-1α, thereby reducing the extent of hypoxia [50][62]. The mTORC1 pathway is believed to play an important role in the induction of plastic properties and the formation of Th1.17 cells (with a pathogenic phenotype) [51][63]. The PI3K (phosphatidylinositol 3-kinase)-AKT-mTORC1-S6K axis has been described as a positive regulator of Th17 cell differentiation that induces nuclear translocation of the RORyt factor [52][64]. In addition, hypoxia and other cellular disturbances (DNA damage, endoplasmic reticulum stress, energy stress) cause transcription of the DDIT4 gene (DNA damage-induced transcript 4, or REDD1). Recent studies have shown that binding of 1,25 (OH)2D3 to VDR can increase DDIT4 gene expression. Considering that activation of DDIT4 leads to inhibition of mTORC1, their interaction can be regulated by 1,25 (OH)2D3 [53][ 65 ]. At the same time, DDIT4 suppresses the activity of mTORC1, inducing the TSC1 / 2 complex, and regulates the production of IL-17 in patients with multiple sclerosis [54][ 72 ]. Under pathological conditions, adenosine triphosphate (ATP) is released from intracellular stores into the extracellular space, where it acts as a stress signal (alarm, warning, DAMP) by binding to purinergic receptors. The purinergic receptor P2X7 (P2X7R), an extracellular ATP-dependent channel, is involved in the secretion of proinflammatory cytokines (which trigger the inflammatory response), cell death, and autophagy [ [29][55][56]38 , 73 , 74 ].