Recent developments in the synthesis of corroles and hexaphyrins are reviewed, highlighting their potential application in photodynamic therapy.
- corroles
- hexaphyrins
- photodynamic therapy (PDT)
- photosensitizer dye
Corroles and hexaphyrins are porphyrinoids with great potential for diverse applications. Like porphyrins, many of their applications are based on their unique capability to interact with light, i.e., based on their photophysical properties. Corroles have intense absorptions in the low-energy region of the uv-vis, while hexaphyrins have the capability to absorb light in the near-infrared (NIR) region, presenting photophysical features which are complementary to those of porphyrins. Despite the increasing interest in corroles and hexaphyrins in recent years, the full potential of both classes of compounds, regarding biological applications, has been hampered by their challenging synthesis.
1. Definition
Corroles are tetrapyrrolic macrocycles containing 4 pyrrole units, 3 methine bridges and a direct pyrrole–pyrrole linkage (Figure 1). Therefore, corroles are contracted porphyrinoids with one methine (=CH–) bridge less than porphyrins, which leads to lower symmetry, higher fluorescence quantum yields, lower oxidation potentials and more intense absorption in the low-energy region of the visible spectrum. Despite being less studied than porphyrins over the years, probably due to their challenging synthesis, there was a significant change in this scenario after the synthetic breakthroughs by Gryko, Gross and Paolesse [1][2][3][4][5]. In fact, many efforts have been devoted to the synthesis of corroles in recent years [6][7][8][9]. Their free-bases and metal complexes have been explored in a wide range of applications [10][11], namely as promising drug candidates for prevention and treatment of diverse diseases, such as diabetes, neurodegenerative diseases and cancer [12][13][14][15][16], as antibacterial agents [17] as well as bio-imaging agents [18]. Corroles also found application in the photodynamic inactivation of microorganisms (PDI) [19], sonodynamic therapy[20] and photodynamic therapy (PDT) [21].
Expanded porphyrins [22][23][24] have gained remarkable attention due to their interesting and versatile features, such as diverse p-conjugation pathways owing to flexible structures [25], near- infrared (NIR) absorption/emission [26], facile interconversion between multiple redox states[27], and multi-metal coordination cavities for various divalent and trivalent metal ions (Figure 1) [28]. Beyond the study of these unique properties and their applications, the interest in expanded porphyrins has been focused on exploring the limits to which the classic Hückel definition of aromaticity may be applied. The goal has been to understand what factors endow a fully conjugated macrocycle with characteristics that can be considered aromatic or antiaromatic. Among the known expanded porphyrins that can combine from 5 to 12 pyrrole rings and diverse meso-bridges, hexaphyrins stand-out because these six-pyrrole macrocycles can adopt various conformations and electronic states such as Hückel aromatic, Hückel antiaromatic, Möbius aromatic, Möbius antiaromatic and stable radical states [29][30].
[26]Hexaphyrins(1.1.1.1.1.1) and [28]hexaphyrins(1.1.1.1.1.1), having six meso-bridges, are attractive molecules in view of aromaticity/antiaromaticity with a 26 and 28 p-electronic system, respectively, intense uv-vis absorption and a small HOMO-LUMO gap (Figure 1). [26]Hexaphyrins(1.1.1.1.1.1) can adopt a double-sided rectangular ring conformation with Hückel aromaticity while [28]hexaphyrins(1.1.1.1.1.1) adopt a single-sided twisted ring conformation with Möbius aromaticity. The unique uv-vis-NIR absorption/emission properties of these expanded π conjugated molecular systems offer useful optical nonlinear properties such as two-photon absorption (TPA) [31], which make them a promising class of two-photon absorption chromophores, with potential for a wide range of applications including microscopy, microfabrication, three-dimensional data-storage, optical power limiting, up-converted lasing, localized release of bio-active species, as well as application in photodynamic therapy [32].
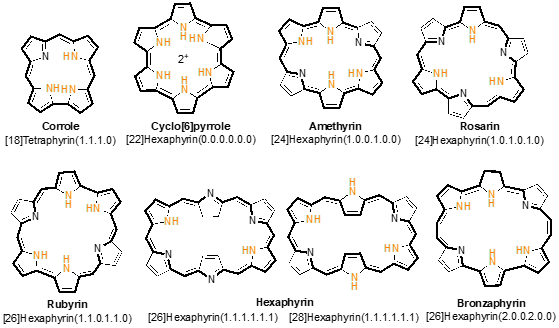
Figure 1. Chemical structures of corrole and hexaphyrins.
2. Synthesis of New Corrole Derivatives
In recent decades, the synthesis of corroles and metallocorroles had attracted considerable attention from the scientific community and some reviews have been published[33][34]. Nevertheless, the relevance of corroles’ applications has driven the development of interesting approaches to construct macrocycles with new moieties, via classical and novel methodologies, collected herein.
2.1. Classical Synthetic Methodologies
One of the most explored routes to meso-substituted corroles relies on the synthesis of bilanes, obtained from the condensation of dipyrromethanes (DPs) with aldehydes, followed by oxidative macrocyclization. This strategy was applied to the synthesis of new corroles to be used in diverse applications.
Liu and coworkers described the synthesis of trans-A2B corroles meso-substituted by phenothiazine (PTZ) and 2,3-di(2-pyridyl)quinoxaline (DPQ) groups. The HCl-catalyzed condensation of PTZ and DPQ formyl derivatives 2 and 4 with 5-pentafluorophenyldipyrromethane (1) followed by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)-promoted macrocyclization gave the target corroles 3 and 5 in 31 and 34% yield, respectively (Scheme 1) [35]. Corroles 3 and 5 are novel donor-acceptor systems, in which the corrole scaffold is an acceptor and PTZ or DPQ moiety is an energy/electron donor. The absorption spectra of both dyads are a superposition of the absorption spectra of the monomers, indicating a weak electronic interaction between the corrole and either PTZ or DPQ groups, which was corroborated by the observed electrochemical properties. A detailed study of the excited state deactivation of both dyads shows that the singlet–singlet energy transfer from PTZ to the corrole unit is the main process in the case of dyad 3, while in the case of 5 a reductive electron transfer, from the ground state of DPQ to the excited state of the corrole, is the main process. Therefore, one dyad mimics the primary process and the other mimics the reaction center events in photosynthesis. The proton-triggered emission studies of dyad 5 and OFET studies indicate that they can be applied in the DNA photocleavage.
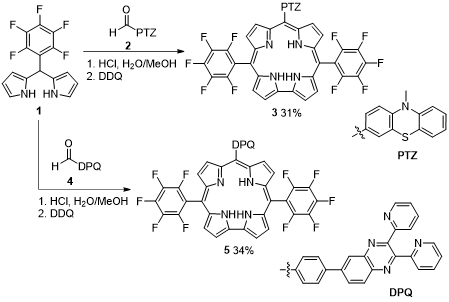
Scheme 1. Synthesis of trans-A2B-corroles bearing phenothiazine and 2,3-di(2-pyridyl)quinoxaline substituents.
The synthesis of 10-(4-methyl-bipyridyl)-5,15-di(pentafluoropheny)corrole (7) was achieved through Gryko’s methodology, i.e., the condensation of dipyrromethane 1 with the bipyridyl-aldehyde 6 in a mixture of H2O/MeOH using HCl as catalyst. Oxidation with DDQ afforded corrole 7 in 17% yield (Scheme 2) [36]. The uv-vis spectrum exhibits a strong Soret band at approximately 400 nm and the Q bands appear between 500 and 700 nm; a single emission band is observed in the fluorescence spectrum. The photophysical properties studied in dichloromethane (DCM) showed that corrole 7 fluorescence quantum yield is 0.04, the internal conversion quantum yield is 0.45 and the triplet quantum yield formation is about 0.54. The singlet oxygen quantum yield, a measure of the ability of the photosensitizer to generate cytotoxic singlet oxygen and an important parameter to evaluate the capability of the molecules to act as photosensitizers for PDT, was measured as 0.47, indicating a high efficiency in energy transfer from the corrole triplet state.
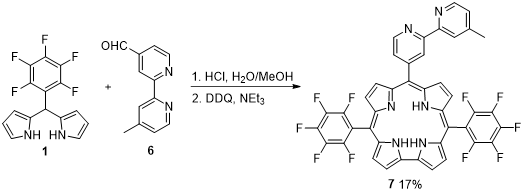
Scheme 2. Synthesis of 10-(4-methyl-bipyridyl)-5,15-di(pentafluorophenyl)corrole.
The trifluoroacetic acid (TFA)-catalyzed condensation of meso-pentafluorophenyl-dipyrromethane (1) with 8-hydroxyquilonine-2-carboxaldehyde (8) gave the low-symmetric A2B-type corrole 9 in 16% yield. Free base corrole 9 was treated with an excess of GaCl3 in refluxing pyridine to produce the Ga(III) corrole 10 in 40% yield (Scheme 3) [37]. According to the fluorescence studies, Ga(III) corrole 10 was a good “off-on-off” pH sensor in 1–14 pH range. The Ga(III) complex presents a fluorescence band with maximum at 603 nm, fluorescence quantum yield of 11.7% and a fluorescence lifetime of 2.42 ns in DCM solution. The intensity of fluorescence decreases gradually with the acidity of the solution and increases when the solution is more basic.
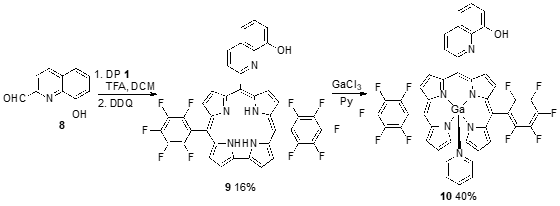
Scheme 3. Synthesis of a 8-hydroxyquinoline-substituted A2B-type free base corrole and the corresponding Ga(III) corrole.
The condensation of 5-(4-nitrophenyl)dipyrromethane (11) with aromatic aldehydes, followed by oxidation with p-chloranil allowed the isolation of trans-A2B corroles 12 in 29–38% yield (Scheme 4) [38]. The photophysical properties in toluene show characteristic absorption/emission spectra with Soret type bands between 420 and 450 nm, Q-type bands around 590–655 nm and the fluorescence bands in the range of 673–690 nm. The fluorinated corroles present fluorescence quantum yields higher than the carboxymethyl derivative, 0.76 for 12a, 0.80 for 12b and 0.77 for 12c. Corroles 12a–d showed an excellent affinity for the fluoride ions, allowing for an unusual selectivity for fluoride ions. The quenching of the fluorescence emission with the addition of fluorine ion indicates that these corroles can be used as fluorine ion sensors.
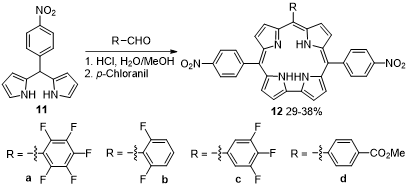
Scheme 4. Synthesis of trans-A2B corroles [where A = 4-nitrophenyl and B = pentafluorophenyl, 2,6-difluorophenyl, 3,4,5-trifluorophenyl and 4-(methoxycarbonyl)phenyl groups].
Liang and coworkers developed the synthesis of three low-symmetry A2B type manganese(III) meso-triarylcorroles providing a push–pull electronic system (Scheme 5). The acid-catalyzed condensation of dipyrromethane 13 with the appropriate aldehyde followed by oxidation with p-chloranil gave corroles 14. The reaction of these macrocycles with manganese chloride in DMF afforded metallocorroles 15a–c in high yields (63–90%) [39]. The fluorescence studies reveal that Mn(III) corroles 15a–c strongly interact with the cell-free circulating tumor DNA in solution and the ability of this interaction increases with the electron-donating character of the 10-substituent of the triarylcorrole. This interaction with DNA can be used for tumor detection and targeting drug delivery in vivo.
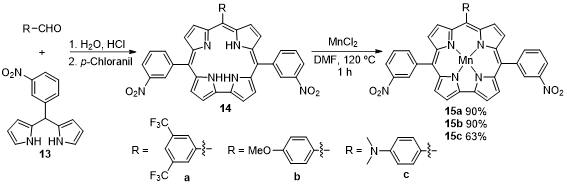
Scheme 5. Synthesis of Mn(III) trans-A2B triarylcorroles [where A = 3-nitrophenyl and B = 4-methoxphenyl, 4-(dimethylamino)phenyl and 4-(trifluoromethyl)2phenyl groups].
Triaryl corroles with a meso-iodoaryl substituent were prepared and their photophysical properties were studied in order to evaluate their capabilities as photosensitizers for PDT. A2B-type triaryl corroles 17 were synthesized through the TFA-catalyzed condensation of DP 1 with 2-hydroxybenzaldehyde and m-iodinated 2-hydroxybenzaldehydes, 16, followed by oxidation with DDQ (Scheme 6) [40]. The presence of iodine atoms did not significantly influence the absorption spectra and only a slight decrease in the absorption coefficient of the Soret band (ca. 420 nm) was observed with the increase in the number of iodine atoms. However, the fluorescence quantum yield and fluorescence lifetime are strongly influenced by the iodine atom. Corrole 17a, without iodine atoms, has a fluorescence quantum yield of 0.143 that decreases to 0.031 in the case of mono-iodo-corrole 17b and to 0.023 for the di-iodo-corrole 17c. The decrease in the fluorescence quantum yield is accompanied by the decrease in the fluorescence lifetime and a slight increase in the triplet quantum yields showcasing the heavy atom effect. Despite the increase in the efficiency of the triplet state formation, the singlet oxygen quantum yield (FD) decreased, going from mono-iodo-corrole 17b (FD = 0.9) to di-iodo-corrole 17c (FD = 0.4).
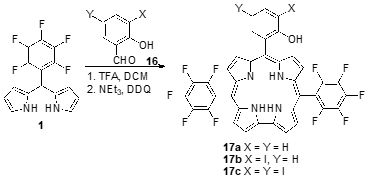
Scheme 6. Iodine-containing A2B-type triaryl corroles synthesized to evaluate the influence of iodine atoms on the photophysical properties.
During the synthesis of the bulky bis-pocket corrole 20a, Chang and coworkers identified another green compound as being the β-chlorinated corrole 20b, fully supported by characterization data, including X-ray crystallography analysis (Scheme 7) [41]. The β-chloro-corrole 20b was formed through the mono-chlorination of corrole 20a in which DDQ acted as both oxidant and chlorinating agent. The presence of a chlorine atom at the β-position of corrole 20b induced a slight red shift in the absorption and fluorescence emission spectra, together with a significant decrease in fluorescence intensity. Fluorescence lifetimes, as well as the fluorescence quantum yields, are shorter for β-chloro-corrole 20b (2.5 ns) than for corrole 20a (4.2 ns). However, the decrease in the fluorescence did not promote the increase in the capability to generate singlet oxygen, since non-chlorinated corrole 20a demonstrated a higher ability to generate singlet oxygen than the β-chlorinated corrole 20b.
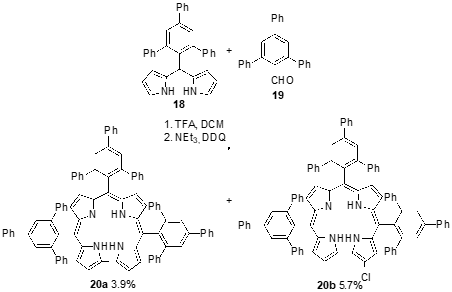
Scheme 7. Synthesis of bulky bis-pocket corroles.
The three component one-pot reaction of pyrrole, p-trifluoromethylbenzaldehyde and azulene (21), catalyzed by TFA, followed by oxidation with DDQ, allowed the isolation of novel meso-triarylazulicorrole 22 (Scheme 8) [42]. Despite the poor yield (<1%), carbacorrole 22 could be converted into the corresponding Cu(III) and Au(III) complexes. Single-crystal X-ray structures were obtained for the free-base and Cu(III) derivatives, uncovering detailed structural information on these novel macrocyles. The meso-triarylazulicorroles show absorption spectra with absorption features extending into the NIR region, opening the way to their application in bioimaging and photodynamic therapy.
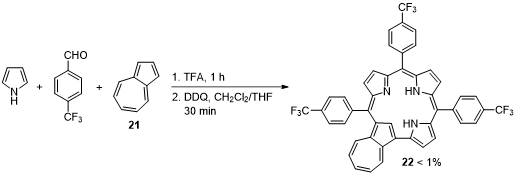
Scheme 8. Synthesis of meso-tri-(p-trifluoromethyl)phenylazulicorrole.
2.2. New Synthetic Methodologies
The above-described syntheses of corroles were based on classical methodologies involving acid-catalyzed condensation of pyrroles or meso-substituted dipyrromethanes and aldehydes, followed by oxidation. Recently, Pinho e Melo et al. described an innovative synthesis of A2B-type corroles, bearing an oxime functionality, by exploring the reactivity of dipyrromethanes toward nitrosoalkenes (Scheme 9) [43]. In situ dehydrohalogenation of the α,α-dihalo-oximes 23 generates transient α-chloro-nitrosoalkenes, which react with dipyrromethanes either via hetero-Diels−Alder reaction or conjugated addition to give the corresponding alkylated dipyrromethanes. The side chains of these DPs undergo another dehydrohalogenation to afford new nitrosoalkenes, allowing the synthesis of the target bilanes 24 (16–40%) upon reaction with a second molecule of the dipyrromethane. The oxidative macrocyclization with DDQ affords the corresponding corroles 25 in 40–84% yields. These are the first examples of meso-substituted corroles with an oxime functionality.
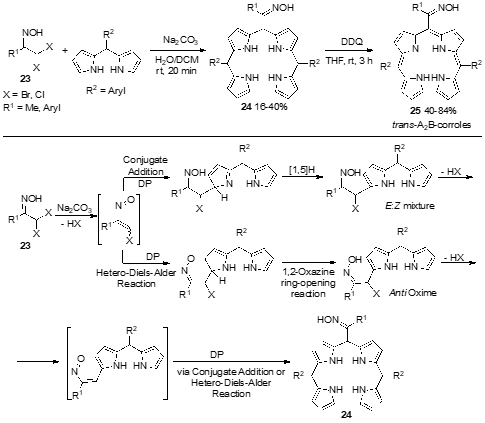
Scheme 9. Synthesis of oxime-containing trans-A2B-corroles based on the reactivity of nitrosoalkenes toward dipyrromethanes.
63. Hexaphyrins: Synthesis and Applications
Gossauer et al. described, in 1983, the first synthesis of hexaphyrins with six meso-bridges, b-substituted aromatic macrocycles with 26-electron conjugation, following a “3 + 3” approach, i.e., through the condensation of two tripyrromethanes [44]. Following this work, meso-substituted hexaphyrins were synthetized using the classical methodologies for the synthesis of porphyrins, such as Lindsey and Rothemund synthetic approaches [45][46]. Although these methods are convenient and effective, when stabilized with electron-withdrawing groups at the meso-position, the simultaneous formation of every size of expanded porphyrins, at least up to dodecamer, inevitably causes serious separation difficulties. Therefore, more ring-size selective syntheses using dipyrromethanes and tripyrromethanes as the starting materials were developed [47][48].
More recently, Osuka and coworkers described the synthesis of hexaphyrins bearing two benzoyl groups at meso-positions and their complexes [49]. Hexaphyrin 103 was synthesized as a stable compound in 12% overall yield by p-toluenesulfonic acid catalyzed cross-condensation of tripyrrane 101 with phenylglyoxal monohydrate (102), at 0 °C under N2 atmosphere with protection from room light for 1.5 h, followed by oxidation with DDQ (Scheme 37). X-Ray diffraction analysis has revealed that 103 adopts a dumbbell-like conformation, held by effective intramolecular hydrogen bonding between the pyrrolic NH protons and benzoyl carbonyl groups. Oxidation of 103 with MnO2 afforded hexaphyrin 104, quantitatively. However, hexaphyrin 104 is easily reduced back to 103 under ambient conditions, indicating that hexaphyrin is more stable than hexaphyrin despite its distinct antiaromatic character. The complexation of 103 with Au(III) afforded two bis-Au(III) isomeric complexes 105a and 105b in 32% and 3%, respectively (Scheme 37). X-Ray crystal analysis showed that both complexes present rectangular conformations with hydrogen bonding between the benzoyl groups and the outer NH protons.
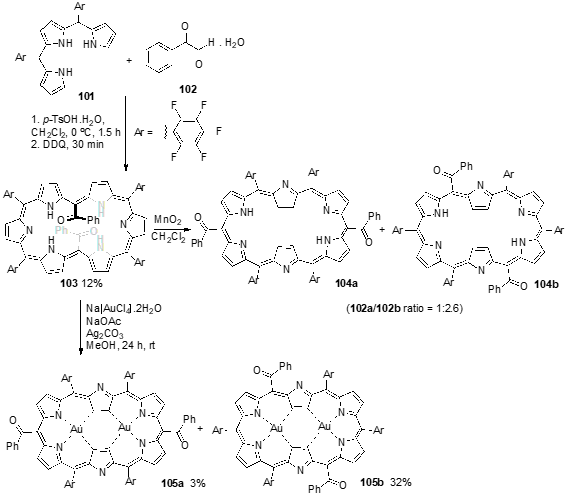
Scheme 10. Synthesis of isomeric 5,20-dibenzoylhexaphyrins and their Gold(III) complexes.
Complex 105b was quantitatively reduced to hexaphyrin Au(II) complex 106 with NaBH4 at room temperature, which could oxidize back to 105b without significant degradation by treatment with MnO2. The benzoyl substituents and pyrrolic NH-protons of the bis-Au(III) complex of [28]hexaphyrin 106 were used as peripheral bidentate coordination sites to prepare the boron(II) complex 107. The complexation was achieved in 90% yield by heating a mixture of 106, BF3-OEt2 and DIPEA in dichloromethane at 40 °C for 24 h (Scheme 38) [50].
The absorption spectrum of the hexaphyrin 103 in CH2Cl2 shows a Soret band at 516 nm and a Q band (without fine structure) between 700–900 nm. The absorption spectra of gold complexes 105a and 105b are similar, showing Soret-like bands around 670 nm and Q-like bands in the NIR region at ca. 822, 930 and 1195 nm, supporting their 26p aromaticity. The absorption spectrum of the reduced complex 106 shows broad absorption bands at 544, 603, and 738 nm and weak absorption in NIR region, reflecting its antiaromaticity. The absorption spectrum of 107 is red-shifted by ca. 100 nm and sharper than that of 106, probably due to structural rigidity and the presence of electron withdrawing BF2-moieties.
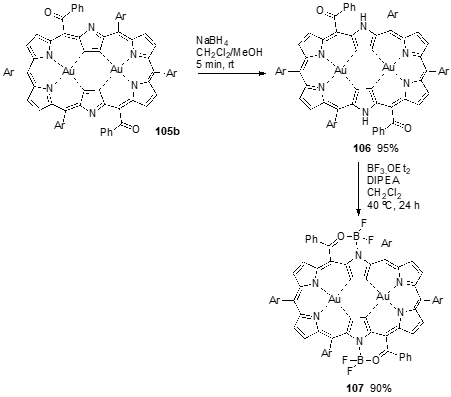
Scheme 11. Synthesis of a [26]hexaphyrin bis-gold(III) complex and the corresponding exocyclic boron complex.
5,20-Bis(ethoxycarbonyl)-[28]hexaphyrin 109 was synthesized using meso-pentafluorophenyl tripyrrane 101 and ethyl 2-oxoacetate (108) as starting materials and the previously described methodology to promote the cross-condensation, followed by oxidation with DDQ (Scheme 39) [51]. X-Ray crystallographic analysis revealed that 109 presents a dumbbell-like conformation held by effective intramolecular hydrogen bonding between the pyrrolic NH protons and carbonyl oxygen of ethoxycarbonyl group. The uv-vis spectrum of 109 displayed characteristic features of antiaromatic porphyrinoids with a broad Soret-like band at 514 nm, and relatively weak Q-like bands. [28]Hexaphyrin 109 can serve as a bis-NNNO ligand to form square planar bis-Ni(II) and bis-Cu(II) complexes. The reaction of 109 with 20 equiv. of nickel(II) acetylacetonate (Ni(acac)2) in dry toluene at 100 °C for 48 h afforded a bis-Ni(II) complex 110 in 48% yield. X-Ray crystallographic analysis has confirmed that 110 has a square planar geometry formed through the coordination of the three nitrogen atoms inner and the carbonyl oxygen atom to the Ni(II) ion, and that this bis-complex kept the dumbbell-like conformation. The complexation with a large excess of CuCl2 in the presence of NaOAc in CH2Cl2, at room temperature, for 24 h, yields the bis-Cu(II) complex 111 in 79% yield, which also takes on a dumbbell-like conformation and square planar geometry. The absorption spectrum of 110 in CH2Cl2 showed a broad Soret-like band at 508 and 621 nm, Q-like band at 830 nm and a weak absorption tail up to 1700 nm. The complexation with copper modified the absorption spectrum of 111, narrowing the Soret-like band at 520 and 635 nm, blue-shifting the Q-like bands to 814 nm and also the very weak absorption tail up to ca. 1500 nm. Oxidation of 5,20-bis(ethoxycarbonyl)-[28]hexaphyrin 109 with a large excess of PbO2 in CH2Cl2 at room temperature for 3 min afforded [26]hexaphyrin 112 in 73% yield. This hexaphyrin was easily reduced back to 109 with NaBH4 in a mixture of CH2Cl2/MeOH at room temperature for 10 min (Scheme 39). Interestingly, the hexaphyrin 112 was the sole product, in contrast with the formation of two isomeric [26]hexaphyrins in the oxidation of 5,20-dibenzoyl-[28]hexaphyrin with MnO2 [52]. The uv-vis-NIR absorption spectrum of 112 showed a sharp Soret-like band at 562 nm and Q-like bands at 712, 770, 892, and 1022 nm, characteristic features of aromatic porphyrinoids.
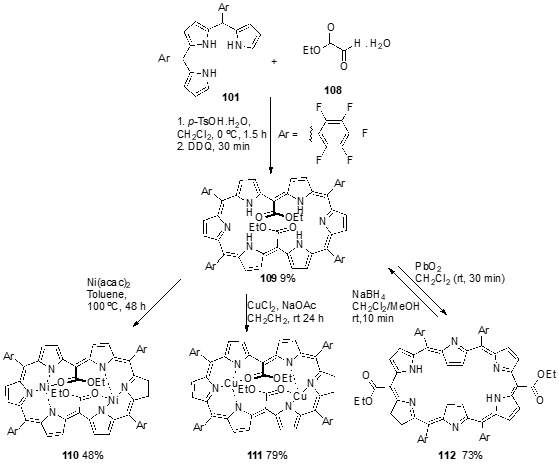
Scheme 12. Synthesis of a 5,20-bis(ethoxycarbonyl)-[28]hexaphyrin, its bis-Ni(II) and bis-Cu(II) complexes and the corresponding 5,20-bis(ethoxycarbonyl)-[26]hexaphyrin.
Following studies on the complexation of aromatic electron-deficient meso-pentafluorophenyl [26]hexaphyrin and electron rich meso-pentafluorophenyl [28]hexaphyrin with Möbius aromaticity, Osuka and coworkers reported the synthesis of π-ruthenium complexes [53]. The treatment of mono- and bis-gold(III) complexes of [28]hexaphyrin 113a and 113b with five equivalents of [RuCl2(p-cymene)] in the presence of NaOAc in a mixture of toluene/DMF at 100 °C for 65 h afforded π-ruthenium complexes 114a and 114b in 45% and 56% yield, respectively (Scheme 40). When the meso-pentafluorophenyl [26]hexaphyrin was submitted to the same reaction conditions, only decomposition products were obtained, suggesting that the formation of ruthenium π-complexes might need an electron-rich hexaphyrin. The absorption spectrum of 114b shows two intense bands at 490 and 656 nm and two less intense and more broad bands at 1066 and 1250 nm. The treatment with TFA yielded the trifluoroacetate 115, with the absorption spectrum of this salt presenting the same bands, and blue shifted to 455, 609, 1066 and 1097 nm.
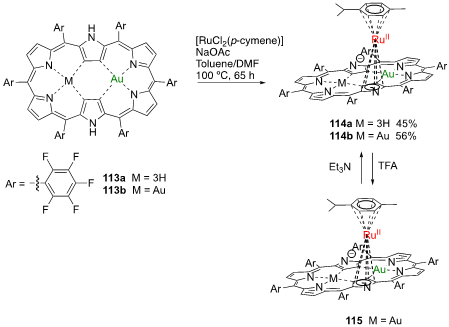
Scheme 13. Synthesis of Ru(II) complexes of meso-pentafluorophenyl-[28]hexaphyrin mono- and bis-Au(III) complexes.
Ru-metalation of [26]hexaphyrin bis-Pd(II) complex 116, a unique complex with a characteristic conjugated aromatic circuit and relatively acidic outer pyrrolic NH protons [54], was achieved through a reaction with [RuCl2(p-cymene)] in the presence of NaOAc in a mixture of toluene/DMF (4:1) at 100 °C for 12 h [55]. The [26]hexaphyrin bis-Pd(II)-bis-Ru(II) complex 117, a triple-decker complex with both the [(p-cymene)Ru(II)] moieties located above and below the center of the hexaphyrin framework, was obtained in 57% yield (Scheme 41). The absorption spectrum of 117 in CH2Cl2 shows two Soret-like bands at 461 and 650 nm and a Q-like band at 829 nm.
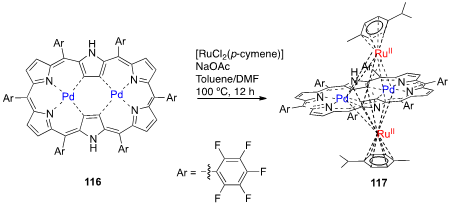
Scheme 14. Synthesis of Ru(II) complexes of meso-pentafluorophenyl-[28]hexaphyrin bis-Pd(II) complex.
Hexaallyloxy-appended hexaphyrin 119 prepared through nucleophilic aromatic substitution reaction of 5,10,15,20,25,30-hexakis(pentafluorophenyl)-[26]hexaphyrin (118) with allyl alcohol, [56] was subjected to olefin metathesis reaction using the improved Hoveyda–Grubbs catalyst. When the reaction was conducted at room temperature, only peripherally strapped [26]hexaphyrin monomer 120 was obtained in a 37% yield. Decreasing the amount of catalyst and increasing the reaction temperature to 40 °C led to the formation of 120 as a minor product (9% yield) and cyclophane-type [26]hexaphyrin dimer 121 in 21% yield (Scheme 42). The treatment of dimer 121 with NaBH4 at room temperature for 10 min afforded [28]hexaphyrin dimer 122 in 30% yield, which could be transformed back into 121 by oxidation with MnO2 (Scheme 42) [57]. The uv-vis-NIR absorption spectrum of aromatic hexaphyrin 120 shows a typical sharp Soret-like band at 569 nm and Q-like bands at 718, 908, and 1031 nm. Dimer 121 shows split Soret-like bands at 543 and 577 nm, attributed to the exciton coupling of the two hexaphyrin cores in a stacked geometry,[58] and Q-like bands 724, 899, and 1031 nm. The absorption spectrum of 122 exhibits characteristics of anti-aromatic [28]hexaphyrins, smaller absorption bands at 490, 526, and 571 nm and a weak absorption at the near-infrared region.
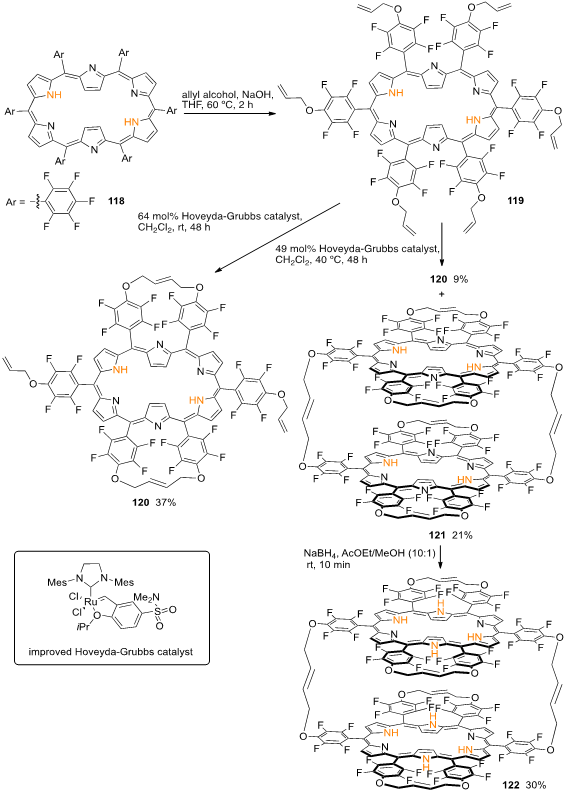
Scheme 15. Synthesis of monomeric and dimeric strapped hexaphyrins.
Following the pioneer work of Osuka and coworkers on regioselective nucleophilic substitution reactions of meso-hexakis(pentafluorophenyl)-substituted [26]hexaphyrin with an alkoxides and isopropyl amine [57], Wiehe and coworkers achieved the synthesis of hexa-glycosylated [28]hexaphyrin 124 in high yield (Scheme 43) [58]. [28]Hexaphyrin 123, obtained in 96% yield through a reduction in [26]hexaphyrin 118 with NaBH4 in methanol at room temperature for 20 min, was transformed into the hexa-glycosilated derivative in 78% yield by nucleophilic substitution of the para fluorine substituent of all six pentafluorophenyl units, using 1-thio-β-D-glucose sodium salt as nucleophile, in dry DMF overnight at room temperature. Attempts to promote the glycosylation of [26]hexaphyrin 118 delivered a complex mixture with traces of the expected product and evidence for the formation of the corresponding reduced form, [28]hexaphyrin 123.
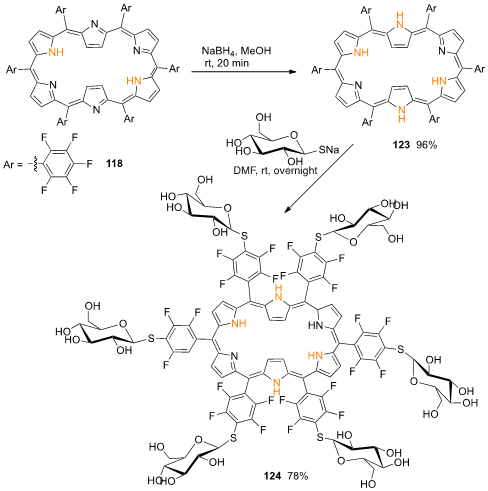
Scheme 16. Synthesis of a hexa-glycosylated [28]hexaphyrin.
To create well-defined NIR-II dyes, chemical modification of the hexapyrrolic core is an alternative viable approach. Recently, Sessler and coworkers reported the synthesis of a new hexaphyrin, pyrihexaphyrin (0.1.0.0.1.0) 125, (Figure 4) that, upon uranyl dication complexation, undergoes ring contraction, affording 22 p-electron aromatic contracted pyrihexaphyrin (0.0.0.0.1.0)-uranyl complex, 126, which displays Hückel type aromatic features. In fact, uv-vis spectroscopic analysis revealed marked differences in comparison to the precursor. A relatively weak Soret-like band was observed at 549 nm along with Q-type bands at 734 and 1003 nm [59]. A robust bis-rhodium(I) complex of p-extended planar, anti-aromatic meso-pentafluorophenyl-b,b-phenylene-bridged hexaphyrin [1.0.1.0.1.0], rosarin 127, was synthesized in 60% yield from the corresponding free-base (Figure 4). Both the ligand and the bis-Rh(I) complex show uv-vis spectra in CH2Cl2 with broad bands between 400–700 nm [58].
The synthesis of doubly N-confused dioxohexaphyrins 128a,b as well as their Zn and Cu complexes, was also reported (Figure 4). These are interesting small molecule-based photoacoustic agents that respond to NIR-II light excitation and constitute a promising platform for deep-tissue NIR-II photoacoustic imaging applications [60].
Hexaphyrin (2.1.2.1.2.1), also known as rubyrins, 129a and b and their rhodium, zinc and copper metal complexes, were obtained to explore the control of aromaticity and cis-/trans-isomeric structure of non-planar hexaphyrins (Figure 4). While the free-base and Zn complex exhibit uv-vis spectra similar to the ones observed for porphyrins, the copper complex has a significant red-shifted bands with Q-like bands at 859 and 947 nm. The rhodium complex displayed a sharp, intense Soret-like band, and a weak Q-like band in the NIR region [61][62].
The incorporation of chalcogenic elements into five-membered rings (i.e., furan and thiophene) significantly affects the dipole vector and charge displacement. This causes a reversal in the direction of the dipole moment between the furan/thiophene and pyrrole [63]. The change in the polarization results in electronic perturbations of the p conjugation. In this regard, chalcogen-substituted hexaphyrins have been synthesized [64][65][66][67][68][69], some of them having been explored as NIR photodynamic therapy agents [70], and others such as dithiabronzaphyrin 130 [69] show intense absorption and fluorescence in the NIR region, opening the way for their use in biological applications.
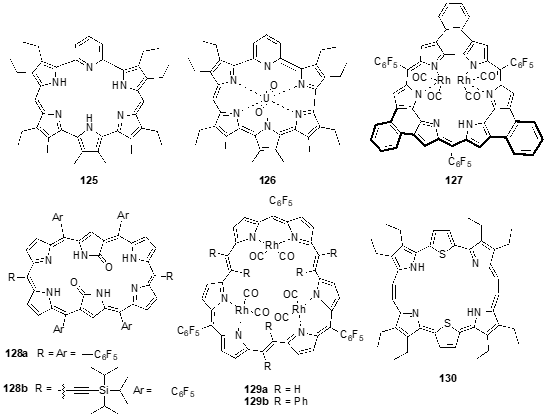
Figure 4. Other hexaphyrins derivatives and analogs synthesized in recent years.
References
- Paolesse, R.; Jaquinod, J.; Nurco, D.J.; Mini, S.; Sagone, F.; Boschi, T.; Smith, K.M. 5,10,15-Triphenylcorrole: A product from a modified Rothemund reaction. Chem. Commun. 1999, 14, 1307–1308, doi:10.1039/A903247I.
- Gross, Z.; Galili, N.; Saltsman, I. The First Direct Synthesis of Corroles from Pyrrole. Angew. Chem. Int. Ed. 1999, 38, 1427–1429, doi:10.1002/(SICI)1521-3773(19990517)38:10<1427::AID-ANIE1427>3.0.CO;2-1.
- Gryko, D.T.; Jadach, K. A Simple and Versatile One-Pot Synthesis of meso-Substituted trans-A2B-Corroles. J. Org. Chem. 2001, 66, 4267–4275, doi:10.1021/jo010146w.
- Teo, R.D.; Hwang, J.Y.; Termini, J.; Gross, Z.; Gray, H.B. Fighting Cancer with Corroles. Chem. Rev. 2017, 117, 2711–2729, doi:10.1021/acs.chemrev.6b00400.
- Jiang, X.; Liu, R.-X.; Liu, H.-Y.; Chang, C.K. Corrole-based photodynamic antitumor therapy. J. Chin. Chem. Soc. 2019, 66, 1090–1099, doi:10.1002/jccs.201900176.
- Barata, J.F.B.; Neves, M.G.P.M.S.; Faustino, M.A.F.; Tomé, A.C.; Cavaleiro, J.A.S. Strategies for Corrole Functionalization. Chem. Rev. 2017, 117, 3192–3253, doi:10.1021/acs.chemrev.6b00476.
- Ghosh, A. Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev. 2017, 117, 3798–3881, doi:10.1021/acs.chemrev.6b00590.
- Orłowski, R.; Gryko, D.; Gryko, D.T. Synthesis of Corroles and Their Heteroanalogs. Chem. Rev. 2017, 117, 3102–3137, doi:10.1021/acs.chemrev.6b00434.
- Sarma, T.; Panda, P.K. Annulated Isomeric, Expanded, and Contracted Porphyrins. Chem. Rev. 2017, 117, 2785–2838, doi:10.1021/acs.chemrev.6b00411.
- Habermeyer, B.; Guilard, R. Some activities of PorphyChem illustrated by the applications of porphyrinoids in PDT, PIT and PDI. Photochem. Photobiol. Sci. 2018, 17, 1675–1690, doi:10.1039/C8PP00222C.
- Nardis, S.; Mandoj, F.; Stefanelli, M.; Paolesse, R. Metal complexes of corrole. Coord. Chem. Rev. 2019, 388, 360–405, doi:10.1016/j.ccr.2019.02.034.
- Haber, A.; Angel, I.; Mahammed, A.; Gross, Z. Combating diabetes complications by 1-Fe, a corrole-based catalytic antioxidant. J. Diabetes Complicat. 2013, 27, 316–321, doi:10.1016/j.jdiacomp.2013.02.005.
- Haber, A.; Gross, Z. Catalytic antioxidant therapy by metallodrugs: Lessons from metallocorroles. Chem. Commun. 2015, 51, 5812–5827, doi:10.1039/C4CC08715A.
- Haber, A.; Mahammed, A.; Fuhrman, B.; Volkova, N.; Coleman, R.; Hayek, T.; Aviram, M.; Gross, Z. Amphiphilic/Bipolar Metallocorroles at Catalyze the Decomposition of Reactive Oxygen and Nitrogen Species, Rescue Lipoproteins from Oxidative Damage, and Attenuate Atherosclerosis in Mice. Angew. Chem. Int. Ed. 2008, 47, 7896–7900, doi:10.1002/anie.200801149.
- Kupershmidt, L.; Okun, Z.; Amit, T.; Mandel, S.; Saltsman, I.; Mahammed, A.; Bar-Am, O.; Gross, Z.; Youdim, M.B.H. Metallocorroles as cytoprotective agents against oxidative and nitrative stress in cellular models of neurodegeneration. J. Neurochem. 2010, 113, 363–373, doi:10.1111/j.1471-4159.2010.06619.x.
- Soll, M.; Bar Am, O.; Mahammed, A.; Saltsman, I.; Mandel, S.; Youdim, M.B.H.; Gross, Z. Neurorescue by a ROS Decomposition Catalyst. ACS Chem. Neurosci. 2016, 7, 1374–1382, doi:10.1021/acschemneuro.6b00144.
- Cardote, T.A.F.; Barata, J.F.B.; Amador, C.; Alves, E.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Cunha, A.; Almeida, A.; Faustino, M.A.F. Evaluation of meso-substituted cationic corroles as potential antibacterial agents. An. Acad. Bras. Cienc. 2018, 90, 1175–1185, doi:10.1590/0001-3765201820170824.
- Liang, X.; Mack, J.; Zheng, L.-M.; Shen, Z.; Kobayashi, N. Phosphorus(V)-Corrole: Synthesis, Spectroscopic Properties, Theoretical Calculations, and Potential Utility for in Vivo Applications in Living Cells. Inorg. Chem. 2014, 53, 2797–2802, doi:10.1021/ic402347w.
- Bornhütter, T.; Shamali, N.; Saltsman, I.; Mahammed, A.; Gross, Z.; Däschlein, G.; Röder, B. Singlet oxygen luminescence kinetics under PDI relevant conditions of pathogenic dermatophytes and molds. J. Photochem. Photobiol. B 2018, 178, 606–613, doi:10.1016/j.jphotobiol.2017.12.015.
- Sharma, V.K.; Mahammed, A.; Soll, M.; Tumanskii, B.; Gross, Z. Corroles and corrole/transferrin nanoconjugates as candidates for sonodynamic therapy. Chem. Commun. 2019, 55, 12789–12792, doi:10.1039/C9CC06494J.
- Mahammed, A.; Gross, Z. Corroles as triplet photosensitizers. Coord. Chem. Rev. 2019, 379, 121–132, doi:10.1016/j.ccr.2017.08.028.
- Alka, A.; Shetti, V.S.; Ravikanth, M. Coordination chemistry of expanded porphyrins. Coord. Chem. Rev. 2019, 401, 213063, doi:10.1016/j.ccr.2019.213063.
- Brewster, J.T.; Zafar, H.; Root, H.D.; Thiabaud, G.D.; Sessler, J.L. Porphyrinoid f-Element Complexes. Inorg. Chem. 2020, 59, 32–47, doi:10.1021/acs.inorgchem.9b00884.
- Jasat, A.; Dolphin, D. Expanded Porphyrins and Their Heterologs. Chem. Rev. 1997, 97, 2267–2340, doi:10.1021/cr950078b.
- Basumatary, B.; Ramana Reddy, R.V.; Rahul Sankar, J. The Curious Case of a Parasitic Twin of the Corroles. Angew. Chem. Int. Ed. 2018, 57, 5052–5056, doi:10.1002/anie.201801555.
- Saito, S.; Osuka, A. Expanded Porphyrins: Intriguing Structures, Electronic Properties, and Reactivities. Angew. Chem. Int. Ed. 2011, 50, 4342–4373, doi:10.1002/anie.201003909.
- Inoue, M.; Osuka, A. Redox‐Induced Palladium Migrations that Allow Reversible Topological Changes between Palladium(II) Complexes of Möbius Aromatic [28]Hexaphyrin and Hückel Aromatic [26]Hexaphyrin. Angew. Chem. Int. Ed. 2010, 49, 9488–9491, doi:10.1002/anie.201005334.
- Rao, Y.; Kim, T.; Park, K.H.; Peng, F.; Liu, L.; Liu, Y.; Wen, B.; Liu, S.; Kirk, S.R.; Wu, L.; et al. π‐Extended “Earring” Porphyrins with Multiple Cavities and Near‐Infrared Absorption. Angew. Chem. Int. Ed. 2016, 55, 6438–6442, doi:10.1002/anie.201600955.
- Stepien, M.; Sprutta, N.; Latos-Grazynski, L. Figure Eights, M bius Bands, and More: Conformation and Aromaticity of Porphyrinoids. Angew. Chem. Int. Ed. 2011, 50, 4288–4340, doi:10.1002/anie.201003353.
- Szyszko, B.; Białek, M.J.; Pacholska-Dudziak, E.; Latos-Grażyński, L. Flexible Porphyrinoids. Chem. Rev. 2017, 117, 2839–2909, doi:10.1021/acs.chemrev.6b00423.
- Ahn, T.K.; Kwon, J.H.; Kim, D.Y.; Cho, D.W.; Jeong, D.H.; Kim, S.K.; Suzuki, M.; Shimizu, S.; Osuka, A.; Kim, D. Comparative Photophysics of [26]- and [28]Hexaphyrins(1.1.1.1.1.1): Large Two-Photon Absorption Cross Section of Aromatic [26]Hexaphyrins(1.1.1.1.1.1). J. Am. Chem. Soc. 2005, 127, 12856–12861, doi:10.1021/ja050895l.
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266, doi:10.1002/anie.200805257.
- Mondal, S.; Naik, P.K.; Adha, J.K.; Kar, S. Synthesis, characterization, and reactivities of high valent metal–corrole (M = Cr, Mn, and Fe) complexes. Coord. Chem. Rev. 2019, 400, 213043, doi:10.1016/j.ccr.2019.213043.
- Ooi, S.; Yoneda, T.; Tanaka, T.; Osuka, A. meso-Free Corroles: Syntheses, Structures, Properties, and Chemical Reactivities. Chem. Eur. J. 2015, 21, 7772–7779, doi:10.1002/chem.201500894.
- Kandhadi, J.; Yan, W.C.; Cheng, F.; Wang, H.; Liu, H.Y. trans-A2B-corrole bearing 2,3-di(2-pyridyl)quinoxaline (DPQ)/phenothiazine moieties: Synthesis, characterization, electrochemistry and photophysics. New J. Chem. 2018, 42, 9987–9999, doi:10.1039/c8nj00606g.
- Pivetta, R.C.; Auras, B.L.; Souza, B.d.; Neves, A.; Nunes, F.S.; Cocca, L.H.Z.; Boni, L.D.; Iglesias, B.A. Synthesis, photophysical properties and spectroelectrochemical characterization of 10-(4-methyl-bipyridyl)-5,15-(pentafluorophenyl)corrole. J. Photochem. Photobiol. A 2017, 332, 306–315, doi:10.1016/j.jphotochem.2016.09.008.
- Cai, F.; Xia, F.; Guo, Y.; Zhu, W.; Fu, B.; Liang, X.; Wang, S.; Cai, Z.; Xu, H. “Off–on–off” type of selectively pH-sensing 8-hydroxyquinoline-substituted gallium(iii) corrole. New J. Chem. 2019, 43, 18012–18017, doi:10.1039/C9NJ04544A.
- Yadav, O.; Varshney, A.; Kumar, A.; Ratnesh, R.K.; Mehata, M.S. A2B corroles: Fluorescence signaling systems for sensing fluoride ions. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 202, 207–213, doi:10.1016/j.saa.2018.05.051.
- Liang, X.; Fang, J.J.; Li, M.Z.; Chen, Q.Y.; Mack, J.; Molupe, N.; Nyokong, T.; Zhu, W.H. Push-pull type manganese(III)corroles: Synthesis, electronic structures and tunable interactions with ctDNA. J. Porphyr. Phthalocyanines 2017, 21, 751–758, doi:10.1142/s1088424617500778.
- Zhao, F.; Zhan, X.; Lai, S.-H.; Zhang, L.; Liu, H.-Y. Photophysical properties and singlet oxygen generation of meso-iodinated free-base corroles. RSC Adv. 2019, 9, 12626–12634, doi:10.1039/C9RA00928K.
- Ali, A.; Cheng, F.; Wen, W.-H.; Ying, X.; Kandhadi, J.; Wang, H.; Liu, H.-Y.; Chang, C.-K. Case synthesis of a β-chloro bulky bis-pocket corrole: Crystallographic characterization and photophysical properties. Chin. Chem. Lett. 2018, 29, 1888–1892, doi:10.1016/j.cclet.2018.03.006.
- Larsen, S.; McCormick-McPherson, L.J.; Teat, S.J.; Ghosh, A. Azulicorrole. ACS Omega 2019, 4, 6737–6745, doi:10.1021/acsomega.9b00275.
- Lopes, S.M.M.; Pinho e Melo, T.M.V.D. Meso-Substituted Corroles from Nitrosoalkenes and Dipyrromethanes. J. Org. Chem. 2020, 85, 3328–3335, doi:10.1021/acs.joc.9b03151.
- Carbon-linked hexapyrrolic systems and heteroatom analogs. In Expanded, Contracted & Isomeric Porphyrins; Jonathan, L., Sessler, S.J.W., Eds.; Tetrahedron Organic Chemistry Series; Elsevier Ltd, ScienceDirect 1997; volume 15, pp. 329–367.
- Neves, M.G.P.M.S.; Martins, R.M.; Tomé, A.C.; Silvestre, A.J.D.; Silva, A.M.S.; Félix, V.; Drew, M.G.B.; Cavaleiro, J.A.S. meso-Substituted expanded porphyrins: New and stable hexaphyrins. Chem. Commun. 1999, 385–386, doi:10.1039/A808952C.
- Shin, J.-Y.; Furuta, H.; Yoza, K.; Igarashi, S.; Osuka, A. meso-Aryl-Substituted Expanded Porphyrins. J. Am. Chem. Soc. 2001, 123, 7190–7191, doi:10.1021/ja0106624.
- Taniguchi, R.; Shimizu, S.; Suzuki, M.; Shin, J.-Y.; Furuta, H.; Osuka, A. Ring size selective synthesis of meso-aryl expanded porphyrins. Tetrahedron Lett. 2003, 44, 2505–2507, doi:10.1016/S0040-4039(03)00328-9.
- Suzuki, M.; Osuka, A. Improved Synthesis of meso-Aryl-Substituted [26]Hexaphyrins. Org. Lett. 2003, 5, 3943–3946, doi:10.1021/ol035650x.
- Ishida, S.-I.; Soya, T.; Osuka, A. A Stable Antiaromatic 5,20-Dibenzoyl [28]Hexaphyrin(1.1.1.1.1.1): Core AuIII Metalation and Subsequent Peripheral BIII Metalation. Angew. Chem. Int. Ed. 2018, 57, 13640–13643, doi:10.1002/anie.201808513.
- Nakai, A.; Kim, J.; Kim, D.; Osuka, A. 5,20-Bis(ethoxycarbonyl)-Substituted Antiaromatic [28]Hexaphyrin and Its Bis-NiII and Bis-CuII Complexes. Chem. Asian J. 2019, 14, 968–971, doi:10.1002/asia.201900189.
- Nakai, A.; Ishida, S.-I.; Soya, T.; Osuka, A. π-Ruthenium Complexes of Hexaphyrins(1.1.1.1.1.1): A Triple-Decker Complex Bearing Two Ruthenoarene Units. Angew. Chem. Int. Ed. 2019, 58, 8197–8200, doi:10.1002/anie.201903212.
- Yoneda, T.; Osuka, A. Synthesis of a [26]Hexaphyrin Bis‐PdII Complex with a Characteristic Aromatic Circuit. Chem. Eur. J. 2013, 19, 7314–7318, doi:10.1002/chem.201301030.
- Yoneda, T.; Kim, T.; Soya, T.; Neya, S.; Oh, J.; Kim, D.; Osuka, A. Conformational Fixation of a Rectangular Antiaromatic [28]Hexaphyrin Using Rationally Installed Peripheral Straps. Chem. Eur. J. 2016, 22, 4413–4417, doi:10.1002/chem.201600262.
- Nakai, A.; Yoneda, T.; Ishida, S.-i.; Kato, K.; Osuka, A. Aromatic and Antiaromatic Cyclophane-type Hexaphyrin Dimers. Chem. Asian J. 2019, 14, 256–260, doi:10.1002/asia.201801751.
- Samala, S.; Dutta, R.; He, Q.; Park, Y.; Chandra, B.; Lynch, V.M.; Jung, Y.M.; Sessler, J.L.; Lee, C.-H. A robust bis-rhodium(I) complex of π-extended planar, anti-aromatic hexaphyrin[1.0.1.0.1.0]. Chem. Commun. 2020, 56, 758–761, doi:10.1039/C9CC09221H.
- Kasha, M.; Rawls, H.R.; El-Bayoumi, M.A. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392, doi:10.1351/pac196511030371.
- Suzuki, M.; Shimizu, S.; Shin, J.-Y.; Osuka, A. Regioselective nucleophilic substitution reaction of meso-hexakis(pentafluorophenyl) substituted [26]hexaphyrin. Tetrahedron Lett. 2003, 44, 4597–4601, doi:10.1016/S0040-4039(03)00950-X.
- Klingenburg, R.; Stark, C.B.W.; Wiehe, A. Nucleophilic Thioglycosylation of Pentafluorophenyl-Substituted Porphyrinoids: Synthesis of Glycosylated Calix[n]phyrin and [28]Hexaphyrin Systems. Org. Lett. 2019, 21, 5417–5420, doi:10.1021/acs.orglett.9b01542.
- Brewster, J.T.; Root, H.D.; Mangel, D.; Samia, A.; Zafar, H.; Sedgwick, A.C.; Lynch, V.M.; Sessler, J.L. UO22+-Mediated ring contraction of pyrihexaphyrin: Synthesis of a contracted expanded porphyrin-uranyl complex. Chem. Sci. 2019, 10, 5596–5602, doi:10.1039/c9sc01593k.
- Shimomura, K.; Kai, H.; Nakamura, Y.; Hong, Y.; Mori, S.; Miki, K.; Ohe, K.; Notsuka, Y.; Yamaoka, Y.; Ishida, M.; et al. Bis-Metal Complexes of Doubly N-Confused Dioxohexaphyrins as Potential Near-Infrared-II Photoacoustic Dyes. J. Am. Chem. Soc. 2020, 142, 4429–4437, doi:10.1021/jacs.9b13475.
- Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. Control of Aromaticity and cis-/trans-Isomeric Structure of Non-Planar Hexaphyrin(2.1.2.1.2.1) and Metal Complexes. Angew. Chem. Int. Ed. 2019, 58, 12524–12528, doi:10.1002/anie.201906946.
- Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. [30]Hexaphyrin(2.1.2.1.2.1) as Aromatic Planar Ligand and Its Trinuclear Rhodium(I) Complex. Inorg. Chem. 2018, 57, 9902–9906, doi:10.1021/acs.inorgchem.8b00977.
- Laidig, K.E.; Speers, P.; Streiwieser, A. Origin of depressed dipole moments in fivemembered, unsaturated heterocycles. Can. J. Chem. 1996, 74, 1215–1220, doi:10.1139/v96-136.
- Ajay, J.; Shirisha, S.; Ishida, M.; Ito, K.; Mori, S.; Furuta, H.; Gokulnath, S. Planar Antiaromatic Core-Modified 24π Hexaphyrin(1.0.1.0.1.0) and 32π Octaphyrin(1.0.1.0.1.0.1.0) Bearing Alternate Hybrid Diheterole Units. Chem. Eur. J. 2019, 25, 2859–2867, doi:10.1002/chem.201805861.
- Abdisa Kerayu, B.; Ching, W.-M.; dela Cruz, J.-a.; Hung, C.-H. 2,5-Thienylene-Strapped Bicyclic and Tricyclic Expanded Porphyrins. ChemPlusChem 2019, 84, 810–815, doi:10.1002/cplu.201900013.
- Ambhore, M.D.; Basavarajappa, A.; Anand, V.G. A wide-range of redox states of core-modified expanded porphyrinoids. Chem. Commun. 2019, 55, 6763–6766, doi:10.1039/C9CC02326G.
- Dash, S.; Ghosh, A.; Srinivasan, A.; Suresh, C.H.; Chandrashekar, T.K. Protonation-Triggered Hückel and Möbius Aromatic Transformations in Nonaromatic Core-Modified [30]Hexaphyrin(2.1.1.2.1.1) and Annulated [28]Hexaphyrin(2.1.1.0.1.1). Org. Lett. 2019, 21, 9637–9641, doi:10.1021/acs.orglett.9b03805.
- Gaur, R.; Ambhore, M.D.; Anand, V.G. Non-planar core-modified dibenzi expanded isophlorin. J. Porphyr. Phthalocyan. 2020, 24, 298–302, doi:10.1142/s1088424619501050.
- Kishore, M.V.N.; Panda, P.K. Revisiting the intense NIR active bronzaphyrin, a 26-π aromatic expanded porphyrin: Synthesis and structural analysis. Chem. Commun. 2018, 54, 13135–13138, doi:10.1039/C8CC07752E.
- Tian, J.; Ding, L.; Xu, H.-J.; Shen, Z.; Ju, H.; Jia, L.; Bao, L.; Yu, J.-S. Cell-Specific and pH-Activatable Rubyrin-Loaded Nanoparticles for Highly Selective Near-Infrared Photodynamic Therapy against Cancer. J. Am. Chem. Soc. 2013, 135, 18850–18858, doi:10.1021/ja408286k.