Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Katarzyna Gaweda-Walerych.
Parkin and PINK1 are key regulators of mitophagy, an autophagic pathway for selective elimination of dysfunctional mitochondria. To this date, parkin depletion has been associated with recessive early onset Parkinson’s disease (PD) caused by loss-of-function mutations in the PARK2 gene, while, in sporadic PD, the activity and abundance of this protein can be compromised by stress-related modifications. Intriguingly, research in recent years has shown that parkin depletion is not limited to PD but is also observed in other neurodegenerative diseases—especially those characterized by TDP-43 proteinopathies, such as amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD).
- TDP-43 pathology
- parkin
- mitophagy
- fronto-temporal lobar degeneration (FTLD)
- amyotrophic lateral sclerosis (ALS)
- Parkinson’s disease (PD)
1. Introduction
Frontotemporal dementia (FTD) is the second most common type of early onset dementia under 65 years of age, characterized by atrophy of the frontal and temporal lobes resulting in behavioral and/or language dysfunction [1,2,3][1][2][3]. Amyotrophic lateral sclerosis (ALS; alias motor neuron disease, MND) is an incurable neurodegenerative and neuromuscular disease, with degeneration of motor neurons in the brain and spinal cord which leads to progressive weakness of muscles, and gradual paralysis followed by respiratory failure [4,5,6][4][5][6]. A total of 10% of ALS cases and c.a. 30–40% of frontotemporal lobar degeneration (FTLD) cases are caused by genetic mutations, the remaining cases being sporadic [7]. Due to extensive clinical and genetic overlap, FTLD and ALS are thought to form a continuum of endophenotypes [8], and according to current diagnostic criteria, the term amyotrophic lateral sclerosis–frontotemporal spectrum disorder (ALS–FTSD) is used [9].
The common hallmark of sporadic and the majority of the genetic forms of ALS–FTSD is TDP-43 (transactive response DNA-binding protein 43 kDa) pathology, characterized by TDP-43 protein depletion from the nucleus and its cytoplasmic aggregation [10,11][10][11]. This pathology is present in the brain and spinal cord of 97% of ALS cases and c.a. 45% of FTLD cases [12,13][12][13].
Parkinson’s disease (PD) is a neurodegenerative disease with progressive death of dopaminergic neurons in the substantia nigra, the brain region producing neurotransmitter dopamine (DA). PD is clinically characterized by bradykinesia, resting tremor, and rigidity [14]. Around 15% of patients have genetic forms of the disease due to mutations in PARK(1–23) genes [15,16][15][16].
Mitochondrial dysfunction has been extensively described in sporadic and genetic forms of ALS–FTSD and PD [6]. In particular, the research on mitochondrial dysfunction in genetic PD caused by biallelic PARK2 mutations [17] has gained pace since the discovery of parkin as a key mediator of mitophagy in 2008 [18] (Figure 1). Only two years earlier, in 2006, TDP-43 has emerged as an important protein for neurodegenerative diseases, such as ALS–FTSD, in which parkinsonian symptoms are reported [11,19][11][19] (Figure 1). Further, the observation of decreased parkin in TDP-43 proteinopathies is a relatively recent finding [20,21][20][21] (Figure 1), and its significance in terms of mitochondrial functioning has been hardly investigated. However, since these initial observations (Figure 1) a plethora of new research that has been performed is presented below.
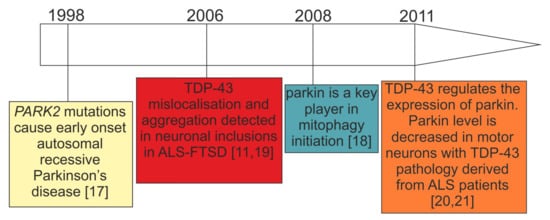
Figure 1. Timeline of the initial key research observations leading to the formation of our working hypothesis: parkin deficit observed in PARK2-related PD and patients with TDP-43 proteinopathies can lead to similar consequences.
2. Parkin and PINK1—The Key Players in Mitophagy Initiation
Mitophagy is a highly coordinated process whereby defective or old mitochondria are eliminated as whole organelles. This process occurs through the lysosomal pathway with the aid of the ubiquitin–proteasome system which concomitantly marks mitochondrial outer membrane proteins for disposal [22,23][22][23]. Mitophagy has been studied in numerous cellular and animal models either as a naturally occurring process or upon induction with various mitochondrial stressors [24]. Among mitophagy inducers, there are agents leading to mitochondrial membrane depolarization, mitochondrial respiratory complex inhibition, mutagenic stress, and proteotoxicity (e.g., CCCP, oligomycin, antimycin A, valinomycin, MPP+, and rotenone) [25,26][25][26].
The first step in mitophagy initiation is performed by mitochondrial kinase PINK1 (PTEN-induced kinase 1) “a sensor of mitochondrial damage” [26]. Under normal conditions, PINK1 expression levels are generally low in cells, because it is cleaved by protease PARL upon translocation to the inner mitochondrial membrane [27[27][28][29],28,29], followed by subsequent ubiquitination and degradation by the proteasome [30]. The other key player is represented by parkin (Parkin RBR E3 Ubiquitin Protein Ligase), which resides predominantly in the cytoplasm. Its expression levels in mitochondria are also normally low, as reviewed in Reference [31].
In response to mitochondrial stress, PINK1 normal processing is hindered. As a consequence, it gets anchored to the translocase of the outer mitochondrial membrane (TOMM) complex where subsequent PINK1 autophosphorylation leads to its auto-activation [32,33][32][33].
Further to this, PINK1 recruits parkin along with ubiquitin onto damaged mitochondria and activates them through phosphorylation on Ser65 [33]. To enhance this process, the Ser65-phosphorylated ubiquitin reinforces parkin activation and stabilization on the OMM [26].
Activated parkin ubiquitinates several mitochondrial outer membrane proteins (e.g., MFN2—mitofusin 2; TOMM20—translocase of outer mitochondrial membrane 20; and VDAC1—voltage dependent anion channel 1), tagging them for proteasome degradation with long ubiquitin chains [26,34][26][34]. Subsequent phosphorylation of these long ubiquitin chains by PINK1 on Ser65 makes it impossible for ubiquitin specific peptidase 30 (USP30) to detach ubiquitin residues from OMM proteins [35]. This generalized polyubiquitination of OMM proteins recruits, in turn, autophagy receptors, such as optineurin (OPTN) and sequestosome-1 (SQSTM1, p62), in such a way as to start forming an autophagosome around the damaged/old mitochondrion, eventually leading to its engulfment and subsequent digestion upon fusion with a lysosome [33].
3. Loss-of-Function (LOF) and Gain-of-Function (GOF) Mechanisms in TDP-43 Proteinopathies
TAR DNA-Binding Protein-43 (TDP-43) was first identified in 2001 as a protein able to bind the HIV-1 TAR binding sequence [61][36], and in 2006, as the main component of aggregates found in the brains of patients with ALS and FTLD [11,19][11][19] (Figure 1). Recently, many reviews have been focused on elucidating the role of this protein in disease and normal development, and, for this reason, the reader is referred to these works for a more detailed description [10,62,63,64,65][10][37][38][39][40]. Briefly, TDP-43 belongs to the class of heterogeneous ribonucleoproteins (hnRNPs) that have been long referred to as the “RNA histones”. Normally, these proteins bind nascent RNA molecules and affect all aspects of RNA processing within the cell, from capping/splicing/polyadenylation to transport/translation and eventually degradation. At the structural level, TDP-43 possesses a highly structured N-terminus domain (NTD) [66,67][41][42] that controls protein dimerization/oligomerization [66,68][41][43]. This NTD is followed by two RNA Recognition Motifs (RRMs) that are responsible for sequence-specific binding to RNA [69,70][44][45] and then by an unstructured C-terminus region that plays a fundamental role in phase separation and aggregation [71,72,73,74][46][47][48][49]. In pathological aggregates, TDP-43 is subject to various post-translational modifications that include ubiquitination, phosphorylation, acetylation, sumoylation and is also cleaved to generate C-terminal fragments [62][37]. From a pathomechanistic point of view, there are two major disease pathways that have been proposed to become disrupted by TDP-43 aggregation and modifications: gain- and loss-of-function disease mechanisms [10]. The gain-of-function mechanisms may include various factors such as direct toxicity of the aggregates [75[50][51][52][53],76,77,78], direct toxicity of the C-terminal fragments [79[54][55],80], or indirect toxicity caused by sequestration of other proteins that are normally in close contact with TDP-43 in the cellular environment [81,82,83,84,85][56][57][58][59][60]. Intriguingly, it is also possible that aggregates might be protective at least during the early stages of the disease. This hypothesis is supported in a recent study based on random mutagenesis of the TDP-43 C-terminus where it has been observed that mutations that increase hydrophobicity and aggregation can decrease toxicity in yeast cells [85][60]. Alternatively, the loss-of-function pathological mechanisms might eventually occur through the sequestration of soluble TDP-43 in the aggregates, thus leaving not enough TDP-43 to perform its normal functions within cells. Therefore, this will result in multiple defects that range from preventing DNA damage to all aspects of RNA processing [86][61]. In support of this view, many recent studies agree that alterations in RNA metabolism could be a major contributor to ALS/FTLD processes in humans [87][62]. Most importantly, it should be kept in mind that all these gain- and loss-of-function possibilities do not necessarily exclude each other, and these different mechanisms could play a role at different stages of the disease. In conclusion, the emerging picture from all of these studies is that, following aggregation of TDP-43, a combination of RNA processing alterations might represent the principal disease contributor in patients with ALS and FTLD [88,89,90][63][64][65].4. Interwoven Relations between TDP-43 and Parkin
Interestingly, apart from Parkinson’s disease, decreased levels of parkin have been found in several TDP-43 proteinopathies (Table 1/Figure 2C). To this date, in fact, there are several examples where the deregulation of parkin expression or its cellular localization has been linked to TDP-43 complex neuropathology and/or has been observed to occur following manipulations of TDP-43 expression levels [20,21,91,92,93,94][20][21][66][67][68][69] (Table 1).Table 1. Effects of TDP-43 pathology, TDP-43 depletion, or overexpression on parkin protein and mRNA levels.
| TDP43-Proteinopathy Model | Cell Type/Treatment Length | Parkin mRNA/Protein |
Accompanying Changes | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| Patients with sporadic ALS (n = 12) vs. control subjects |
ca. 1000 motor neurons/- | Trend for decreased mRNA (microarray) |
- | [95] | [70] | ||||
| Patients with sporadic ALS (n = 11) vs. control subjects (n = 3) |
Spinal cord motor neurons—only those with TDP-43 inclusions/- | Decreased protein (IF) | - | [21] | |||||
| Carriers of | PGRN | mutations from families with FTLD | Human primary skin fibroblasts with | PGRN | mutations | Decreased mRNA/protein by ca. 60% (qRT-PCR) | Unchanged MFN2 and VDAC1 mRNA and protein | [94] | [69] |
| Mouse TDP-43 knockdown | Striatum injection of antisense oligonucleotides/2 weeks | Decreased mRNA by ca. 70% (RNAseq) | - | [20] | |||||
| Mouse TDP-43 knockdown | Brain and spinal-cord injection of antisense oligonucleotides/2 weeks | Decreased mRNA by ca. 80% (qRT-PCR) | - | [21] | |||||
| TDP-43 knockdown in human neurons (TDP-43 expression reduction by 60–75%) | Human neurons (iPSC-derived and HUES6 line) lentiviral shRNA constructs/na | Decreased mRNA by ca. 25% (qRT-PCR) | - | [21] | |||||
| TDP-43 silencing (siRNA) in HEK293T | Human HEK293T (DMSO vs. mitochondrial uncoupler CCCP; siTDP-43 or si CTRL)/na | Decreased protein cytoplasmic localization (IF) | Decreased prohibitin 2 (PHB2) |
[91] | [66] | ||||
| TDP-43 silencing (siRNA) in skin fibroblasts derived from patients with FTLD | Human primary skin fibroblasts with | PGRN | mutations and control fibroblasts (siTDP-43 or siCTRL)/48 h |
Decreased protein by ca. 40% (WB) |
- | [94] | [69] | ||
| Overexpression of wild-type TDP-43-HA or mutant TDP-43-Q331K | Primary mouse neurons/motor cortex and human HEK293T cells/48 h | Decreased endogenous parkin mRNA and protein by c.a. 50% (qRT-PCR, WB) | Increased PINK1 protein | [92] | [67] | ||||
| Exogenous co-expression of wild-type TDP-43-HA and intron-free human parkin or intron-free PINK1 | Human HEK293T cells/48 h | Decreased intron-free parkin mRNA and protein by c.a. 50% (qRT-PCR) |
Increased cleaved PINK1 protein forms insoluble cytoplasmic aggregates | [92] | [67] | ||||
| Transgenic | Drosophila | knock-in of wild-type human TDP-43-H | Fly heads/na | Decreased mRNA and protein by c.a. 45% (qRT-PCR, WB) | - | [92] | [67] | ||
| Wild-type TDP-43 overexpression |
Human HEK293T (DMSO vs. mitochondrial uncoupler CCCP; wild-type pLX-TDP-43-v5 vector/na) |
Increased protein cytoplasmic localization (IF) | Increased prohibitin 2 (PHB2) |
[91] | [66] | ||||
| Wild-type TDP-43 overexpression |
Human primary skin fibroblasts with transiently silenced PGRN (48 h) overexpressing wild-type flag-TDP-43 (24 h) | Increased protein by c.a. 40% (WB) |
Increased PGRN protein | [94] | [69] | ||||
| Wild-type TDP-43 overexpression | Human skin fibroblasts with | PGRN | mutations overexpressing wild-type flag-TDP-43 (48 h) | Decreased protein by c.a. 50% (WB) |
- | [94] | [69] | ||
| Transgenic mouse with heterozygous knock-in of human mutant TDP-43 (A315T) | Whole-brain tissue | mRNA and protein reduced by 70% compared to wild-type controls (qRT-PCR, WB) |
Abnormal neuronal mitochondrial cristae, fusion and fission defects; | [96] | [71] | ||||
| Overexpressed wild-type TDP-43 | Human M17 neuroblastoma cells | Increased protein by c.a. 50% (WB) |
- | [93] | [68] | ||||
| Transgenic mouse with knock-in of human mutant TDP-43A315T | Increased mRNA by c.a. 50% (qRT-PCR) |
- | [93] | [68] |
Abbreviations: IF—immunofluorescence, qRT-PCR—quantitative real-time PCR; iPSC—induced pluripotent stem cells, WB—Western blot. Experiments in Table 1 are presented as follows: dark grey-colored rows—evidence from patients with TDP-43 proteinopathies; light grey-colored rows: experiments with TDP-43 silencing; white smoke-colored rows—experiments with TDP-43 overexpression.
4.1. Consistent Effects of TDP-43 Depletion on Parkin Levels
At the mechanistic level of RNA processing, TDP-43 has turned out to be crucial for the maintenance of brain enriched mRNAs with long introns (>100 kb) such as PARK2 [20,21][20][21]. Indeed, PARK2 pre-mRNA possesses multiple TDP-43 binding sites, suggesting that the control of its stability depends on TDP-43 RNA binding function, at least partially [20] (Figure 2C). In keeping with this view, and irrespectively of animal or cellular models under investigation, TDP-43 depletion consistently resulted in parkin mRNA/protein downregulation [20,21,91,94][20][21][66][69] (Table 1).
To date, a decrease in parkin mRNA has been observed upon TDP-43 depletion in mouse adult brain, stem cell-derived human neurons, HEK293T cells, human primary skin fibroblasts, and motor neurons obtained from patients with sporadic ALS [20,21,91,94][20][21][66][69] (Table 1). However, in motor neurons derived from patients with sporadic ALS, parkin decrease correlated with the presence of TDP-43 aggregates while ca. 95% of motor neurons without TDP-43 pathology showed normal parkin levels [21].
Recently, Sun et al., have elegantly shown that TDP-43 can affect parkin expression post-transcriptionally and in an intron/UTR-independent manner [92][67]. In a recent study, the authors have observed that overexpression of human TDP-43 in HEK293T cells downregulated plasmid-expressed, intron-free parkin, both at the mRNA and protein level (Table 1). This required both the RNA-binding and protein–protein interaction domains of TDP-43 that included the RNA recognition motif 1 (RRM1) and the glycine-rich domain (GRD) domain in the C-terminus [92][67].
In conclusion, the consistency of the results of TDP-43 depletion (Table 1) in different animal and cellular models and in patients with ALS/FTLD suggests that TDP-43 loss-of-function is crucial to maintaining parkin expression levels. Whether this is the only mechanism by which parkin decreases in ALS/FTLD remains elusive.
4.2. Discrepant Effects of TDP-43 Overexpression on Parkin Levels
Supporting the notion of parkin being a direct target for TDP-43, a significant increase in PARK2 mRNA was observed in transgenic mice (hTDP43-Tg) brains compared to controls and a few cellular TDP-43 overexpression models [91,93,94][66][68][69]. Transgenic hemizygous mice harboring human TDP-43 A315T had increased levels of both soluble and insoluble parkin (by ca. 50 and 60%, respectively) [93][68] (Table 1). In contrast, other groups reported parkin downregulation upon wild-type and mutant TDP-43 overexpression, depending on the cellular model used [92,94,96][67][69][71] (Table 1).
Apart from parkin, TDP-43 ectopic expression influenced also other mitophagy key players. Sun et al. reported accumulation of cleaved PINK1(~52 kDa) insoluble aggregates in the cytoplasm via the mechanism of TDP-43 overexpression–related impairment of the proteasomal activity [92][67]. To confirm that these findings hold true also in vivo, the authors confirmed aggregation of cleaved endogenous PINK1 in the motor cortex of mice expressing Q331K mutant of TDP-43 [92][67]. Finally, they demonstrated that TDP-43-driven PINK1 accumulation affected negatively mitochondrial respiratory functions and lifespan in the Drosophila model [92][67].
At present, there is no satisfactory explanation for the observed differences between TDP-43 overexpression experiments. In contrast to TDP-43 knockdown, it should nonetheless be considered that TDP-43 overexpression levels may vary substantially between different labs/experimental systems. At the cellular level, this may result in the overexpressed TDP-43 binding/interacting with different partners, depending on the absolute level of overexpression reached during the study. If this could be the case, it would not be surprising that different overexpression levels could even have opposite effects on parkin expression. Of course, such a possibility would have to be experimentally tested and this is an area that certainly deserves further investigation.
4.3. Parkin as an E3-Ubiquitin Ligase Affects TDP-43 Aggregation
Notwithstanding the fact parkin levels are regulated by TDP-43, it has been shown that parkin, in turn, can affect the state of TDP-43 within cells. In particular, Hebron et al. have shown that parkin promoted Lys-48- and Lys-63-linked ubiquitination of TDP-43 and formed a multi-protein complex with histone deacetylase 6 (HDAC6) inducing sequestration of TDP-43 into cytosolic inclusions [93][68]. Co-expression of exogenous parkin and TDP-43 increased cytosolic co-localization of TDP-43, parkin, and ubiquitin. Moreover, parkin double knockout mice presented significantly higher levels of endogenous TDP-43 than control ones, although the number of hippocampal cells positive for TDP-43 was similar between transgenic and control mice [97][72].
4.4. Unanswered Question 1: Is Parkin Downregulation in TDP-43 Proteinopathies Functionally Relevant (Molecular Biology Perspective)?
While biallelic PARK2 mutations lead to complete parkin depletion in patients with PD [98,99][73][74] (Figure 2A), the parkin decrease in carriers of single PARK2 mutations remains yet to be determined (Figure 2B). In fact, it is important to note that in cellular PD models almost complete parkin deficiency (ca. 80%), obtained by silencing with small interfering RNAs (siRNA), is sufficient to trigger mitochondrial dysfunction in wild-type fibroblasts [100][75]. However, in contrast to patients with EOPD due to homozygous PARK2 mutations, healthy carriers of heterozygous parkin mutation do not present abnormal mitochondrial function, including deregulated ultrastructure, morphology, and metabolism [99,101][74][76]. Likewise, 50% parkin silencing, which models haploinsufficiency (that would correspond to single PARK2 mutation carriers), did not change any mitochondrial parameters, thus indicating the importance of the dose [100][75].
How do the above-mentioned observations correspond to TDP-43 proteinopathies? First of all, many transcriptomic/proteomic analyses in patients with ALS–FTSD or TDP-43 proteinopathy animal models do not report parkin expression alterations [102,103,104,105,106][77][78][79][80][81]. These might be due to the fact that all “omic” results (RNAseq, microarrays, and proteomics) carry a bias, since they are not able to distinguish between healthy cells and those with TDP-43 pathology. Moreover, the severity of TDP-43 pathology may be of varying grades [107,108][82][83].
In models where parkin expression changes are observed (Section 5.1 and Table 1), PARK2 mRNA decrease upon TDP-43 depletion ranges from c.a. 25% in human neurons, 60% in human fibroblasts to 60–80% in rodent models. However, TDP-43 depletion does not mirror the complex pathology present in the brains of patients with ALS–FTLD. Thus, the local threshold effect might be extremely important, in terms of neurons with TDP-43 pathology and magnitude of parkin decrease. Furthermore, additional genetic or epigenetic risk factors may decide to what extent mitochondrial dysfunction would manifest itself.
Since carriers of single PARK2 mutations can present subclinical brain dysfunction (see Section 3.2 for detailed description) it could therefore be speculated that subtle parkin decrease in TDP-43 proteinopathies can lead to similar subclinical phenotypes as in single PARK2 mutation carriers. Nonetheless, this is an assumption that will need careful validation in the future. Indeed, there are limited existing data regarding parkinsonian phenotypes observed in TDP-43 proteinopathies, and this subject is discussed in Section 5.5 below.
4.5. Unanswered Question 2: What Is the Evidence of Parkinsonism in TDP-43 Proteinopathies (Clinical Perspective)?
ALS–FTSD comprises cases with dementia (ALS–FTD), cases with behavioral and/or cognitive impairment without dementia (ALSbci, ALSbi, and ALSci), ALS–parkinsonism/dementia complex (ALS–PDC, named also Western Pacific variant of ALS, lytico bodig), and other mixed variants [9].
ALS–FTSD is predominantly associated with TDP-43 proteinopathy [109][84], ALS being the most common one, and includes many movement disorders presentations, but MND and parkinsonian symptoms predominate.
First of all, it is important to keep in mind that cases of ALS–FTSD that have not been genetically defined may present other types of pathology (such as TAU, FUS, etc.). For this reason, in this review we focus on parkinsonism in genetic forms of ALS–FTSD with confirmed TDP-43 proteinopathy, caused by mutations in C9orf72, PGRN, and TARDBP [110,111,112,113][85][86][87][88].
Regarding FTLD it is interesting to note that the first links between this disease and parkinsonism extend before the recognition of TDP-43 proteinopathies. Indeed, until a few decades ago parkinsonian symptoms were regarded as a rare manifestation of FTLD associated with MAPT mutation, so the term frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) was coined [114][89]. More recently, the association between progranulin mutations and parkinsonism [115,116,117][90][91][92] enabled us to distinguish between FTDP-17 (MAPT) and FTDP-17 (PGRN) [117][92]. Currently, the FTDP-17 term is no longer used clinically, considering that parkinsonism in FTLD has been shown to extend far beyond mutations in chromosome 17 [118][93].
Usually, FTLD is associated with either parkinsonian symptoms (e.g., in PGRN mutations) or with MND (e.g., in C9orf72).
In genetic FTLD parkinsonism is the most frequent movement disorder manifestation. It occurs in about 80% of cases. However, only ~ 1 in 3 cases with C9orf72 mutation and 1 in 10 cases with PGRN mutations present with parkinsonism at onset [119][94]. According to other sources, depending on the population, parkinsonian symptoms occur in >20% of patients with ALS linked to C9orf72 mutation, and as much as 50% to 75% of cases with bvFTD [120,121][95][96].
PGRN mutations are associated predominantly with parkinsonism and only occasionally with MND [122,123][97][98]. In some PGRN kindreds, parkinsonism has been reported in up to 80% of cases [124][99]. Importantly, patients with PGRN mutations demonstrate presynaptic dopaminergic deficit, as evidenced by Dopamine Transporter Scan (DAT-Scan) [125][100]. Unfortunately, DAT-Scan is not routinely performed, and parkinsonism in FTLD is usually described only based on clinical presentation.
It seems that TARDBP mutations may have a broader symptomatic spectrum than other gene mutations in FTLD, as they may be associated with all three conditions: ALS, FTLD, and parkinsonism [126][101].
At the clinical level, parkinsonism in FTLD syndromes is typically characterized by akinetic–rigid phenotype (symmetrical muscle rigidity, bradykinesia, hypokinesia, parkinsonian gait, and rarely resting tremor) [127][102] and may share clinical features with either progressive supranuclear palsy (PSP) or corticobasal syndrome (CBS) [128,129][103][104]. Sometimes multiple system atrophy-like presentations occur with dysautonomia, ataxia, and pyramidal symptoms [127][102].
Akinetic–rigid parkinsonism, as far as TDP-43 proteinopathies are concerned, occurs commonly in PGRN mutations and is rather uncommon in C9orf72 mutations and VCP (Valosin Containing Protein) mutations and rare in TARDP mutations cases. PSP-like features may be rarely observed in both C9orf72 and PGRN mutation carriers, CBS-like features are uncommon in PGRN and rare in C9orf72 [127][102]. Of note, parkinsonian symptoms may be the first symptom in FTLD or develop after the occurrence of language or behavioral symptoms [121,130][96][105].
Among inherited FTLD cases, MND is observed mainly in patients carrying C9orf72 or TARDBP gene mutations, but also those with DCTN1 (Dynactin Subunit 1) and VCP gene mutations.
In some very rare clinical entities, such as Perry disease, caused by DCTN1 mutation, TDP-43 pathology is predominant [131][106]. In others, it co-occurs with tau pathology (PSP, CBD) or alpha-synuclein pathology (PD, DLB) [13]. In Perry disease, parkinsonism, central hypoventilation, and weight loss [131][106] are accompanied by behavioral manifestations. The syndrome shares symptoms of PD, ALS and may fall into the FTLD spectrum [132][107].
Another example of the complex relationship between ALS and parkinsonism is the rare variant of ALS: ALS and parkinsonism/dementia complex (ALS/PDC) in which TDP-43 pathology may be accompanied by alpha-synuclein pathology. In ALS/PDC three types of pathology were described: the tauopathy-dominant type, the TDP-43 proteinopathy-dominant type, and the synucleinopathy-dominant type [133][108].
The frequency of parkinsonism in ALS–FTSD with TDP-43 proteinopathy cannot be easily established. Parkinsonism may be an under-diagnosed phenomenon in ALS–FTSD with TDP-43 proteinopathy due to several reasons. First, genetic testing and/or neuropathological examination is not routinely performed worldwide, remaining unproven in many cases. Secondly, C9orf72 is a relatively recently described mutation as are Strong et al.’s criteria [9]. Third, at the diagnostic stage patients usually attend either a Dementia Clinic or Movement Disorders Clinics. Inevitably, some clinics focus mainly on cognitive/behavioral symptoms or motor symptoms and a mixed presentation of ALS–FTSD may be overlooked, especially if parkinsonism is not present at onset and develops later. At more advanced disease stages when the patients require constant supervision/care they are rarely seen by movement disorder specialists. Therefore, parkinsonism may not be diagnosed even in cases with pronounced akinetic–rigid manifestations. Furthermore, since akinetic–rigid parkinsonism is not a commonly known presentation of parkinsonism, specialists without movement disorder expertise are less likely to diagnose correctly someone that does not present with tremor phenotype.
In conclusion, the theme of parkinsonism in ALS–FTLD guarantees further research since at present there are a lot of gaps that need to be filled, specifically regarding parkinsonism in sporadic forms of ALS–FTSD with defined neuropathology and its clinical subgroups. Finally, it would be interesting to investigate whether parkinsonian symptoms observed in ALS–FTLD could be mediated by TDP-43-associated parkin decrease.
References
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014.
- Pottier, C.; Ravenscroft, T.A.; Sanchez-Contreras, M.; Rademakers, R. Genetics of FTLD: Overview and what else we can expect from genetic studies. J. Neurochem. 2016, 138, 32–53.
- Mann, D.M.A.; Snowden, J.S. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017, 27, 723–736.
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069.
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 2019, 78, 98–110.
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950.
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; McLaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobaGyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174.
- Borroni, B.; Alberici, A.; Buratti, E. Review: Molecular pathology of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2019, 45, 41–57.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95.
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 28.
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178.
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608.
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803.
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611.
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468.
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497.
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14.
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185.
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53.
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248.
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20.
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867.
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712.
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879.
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769.
- Gaweda-Walerych, K.; Zekanowski, C. Integrated pathways of parkin control over mitochondrial maintenance—Relevance to Parkinson’s disease pathogenesis. Acta Neurobiol. Exp. 2013, 73, 199–224.
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101.
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866.
- Pallanck, L.J. Culling sick mitochondria from the herd. J. Cell Biol. 2010, 191, 1225–1227.
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13.
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596.
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293.
- Afroz, T.; Perez-Berlanga, M.; Polymenidou, M. Structural Transition, Function and Dysfunction of TDP-43 in Neurodegenerative Diseases. Chimia Int. J. Chem. 2019, 73, 380–390.
- Buratti, E. Trends in Understanding the Pathological Roles of TDP-43 and FUS Proteins. Adv. Exp. Med. Biol. 2021, 1281, 243–267.
- Francois-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301.
- Mompean, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. The TDP-43 N-terminal domain structure at high resolution. FEBS J. 2016, 283, 1242–1260.
- Qin, H.; Lim, L.Z.; Wei, Y.; Song, J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. USA 2014, 111, 18619–18624.
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Pluckthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45.
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276, 36337–36343.
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449.
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317.
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549.
- Mompean, M.; Chakrabartty, A.; Buratti, E.; Laurents, D.V. Electrostatic Repulsion Governs TDP-43 C-terminal Domain Aggregation. PLoS Biol. 2016, 14, e1002447.
- Pantoja-Uceda, D.; Stuani, C.; Laurents, D.V.; McDermott, A.E.; Buratti, E.; Mompean, M. Phe-Gly motifs drive fibrillization of TDP-43’s prion-like domain condensates. PLoS Biol. 2021, 19, e3001198.
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824.
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE 2014, 9, e86720.
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339.
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176.
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524.
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335.
- Dammer, E.B.; Fallini, C.; Gozal, Y.M.; Duong, D.M.; Rossoll, W.; Xu, P.; Lah, J.J.; Levey, A.I.; Peng, J.; Bassell, G.J.; et al. Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS ONE 2012, 7, e38658.
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; Bowser, R. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012, 124, 717–732.
- Mackenzie, I.R.; Neumann, M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 40–43.
- Shelkovnikova, T.A.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2014, 23, 2298–2312.
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162.
- Vanden Broeck, L.; Callaerts, P.; Dermaut, B. TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71.
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179.
- Hardy, J.; Rogaeva, E. Motor neuron disease and frontotemporal dementia: Sometimes related, sometimes not. Exp. Neurol. 2014, 262 Pt B, 75–83.
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558.
- Solomon, D.A.; Mitchell, J.C.; Salcher-Konrad, M.T.; Vance, C.A.; Mizielinska, S. Review: Modelling the pathology and behaviour of frontotemporal dementia. Neuropathol. Appl. Neurobiol. 2019, 45, 58–80.
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15.
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953.
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115.
- Gaweda-Walerych, K.; Walerych, D.; Berdynski, M.; Buratti, E.; Zekanowski, C. Parkin Levels Decrease in Fibroblasts with Progranulin (PGRN) Pathogenic Variants and in a Cellular Model of PGRN Deficiency. Front. Mol. Neurosci. 2021, 14, 676478.
- Rabin, S.J.; Kim, J.M.; Baughn, M.; Libby, R.T.; Kim, Y.J.; Fan, Y.; La Spada, A.; Stone, B.; Ravits, J. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum. Mol. Genet. 2010, 19, 313–328.
- Stribl, C.; Samara, A.; Trumbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabe de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784.
- Wenqiang, C.; Lonskaya, I.; Hebron, M.L.; Ibrahim, Z.; Olszewski, R.T.; Neale, J.H.; Moussa, C.E. Parkin-mediated reduction of nuclear and soluble TDP-43 reverses behavioral decline in symptomatic mice. Hum. Mol. Genet. 2014, 23, 4960–4969.
- Zilocchi, M.; Colugnat, I.; Lualdi, M.; Meduri, M.; Marini, F.; Corasolla Carregari, V.; Moutaoufik, M.T.; Phanse, S.; Pieroni, L.; Babu, M.; et al. Exploring the Impact of PARK2 Mutations on the Total and Mitochondrial Proteome of Human Skin Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 423.
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 1041–1053.
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.; Smeitink, J.A.; Cookson, M.R.; et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008, 64, 555–565.
- Grunewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.; et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE 2010, 5, e12962.
- Zucca, S.; Gagliardi, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. RNA-Seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data 2019, 6, 190006.
- Briese, M.; Saal-Bauernschubert, L.; Luningschror, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116.
- Andres-Benito, P.; Gelpi, E.; Povedano, M.; Santpere, G.; Ferrer, I. Gene Expression Profile in Frontal Cortex in Sporadic Frontotemporal Lobar Degeneration-TDP. J. Neuropathol. Exp. Neurol. 2018, 77, 608–627.
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausin, K.; Lachen-Montes, M.; Santamaria, E.; Fernandez-Irigoyen, J.; Jerico, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2018, 20, 4.
- Mol, M.O.; Miedema, S.S.M.; van Swieten, J.C.; van Rooij, J.G.J.; Dopper, E.G.P. Molecular Pathways Involved in Frontotemporal Lobar Degeneration with TDP-43 Proteinopathy: What Can We Learn from Proteomics? Int. J. Mol. Sci. 2021, 22, 10298.
- Mackenzie, I.R.; Neumann, M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017, 134, 79–96.
- Tan, R.H.; Kril, J.J.; Fatima, M.; McGeachie, A.; McCann, H.; Shepherd, C.; Forrest, S.L.; Affleck, A.; Kwok, J.B.; Hodges, J.R.; et al. TDP-43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes. Brain 2015, 138, 3110–3122.
- Espay, A.J.; Litvan, I. Parkinsonism and frontotemporal dementia: The clinical overlap. J. Mol. Neurosci. 2011, 45, 343–349.
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273.
- Rowe, J.B. Parkinsonism in frontotemporal dementias. Int. Rev. Neurobiol. 2019, 149, 249–275.
- Park, H.K.; Chung, S.J. New perspective on parkinsonism in frontotemporal lobar degeneration. J. Mov. Disord. 2013, 6, 1–8.
- Rayaprolu, S.; Fujioka, S.; Traynor, S.; Soto-Ortolaza, A.I.; Petrucelli, L.; Dickson, D.W.; Rademakers, R.; Boylan, K.B.; Graff-Radford, N.R.; Uitti, R.J.; et al. TARDBP mutations in Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 312–315.
- Foster, N.L.; Wilhelmsen, K.; Sima, A.A.; Jones, M.Z.; D’Amato, C.J.; Gilman, S. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference Participants. Ann. Neurol. 1997, 41, 706–715.
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919.
- Boeve, B.F.; Baker, M.; Dickson, D.W.; Parisi, J.E.; Giannini, C.; Josephs, K.A.; Hutton, M.; Pickering-Brown, S.M.; Rademakers, R.; Tang-Wai, D.; et al. Frontotemporal dementia and parkinsonism associated with the IVS1 + 1G->A mutation in progranulin: A clinicopathologic study. Brain 2006, 129, 3103–3114.
- Boeve, B.F.; Hutton, M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: Introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch. Neurol. 2008, 65, 460–464.
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534.
- Gasca-Salas, C.; Masellis, M.; Khoo, E.; Shah, B.B.; Fisman, D.; Lang, A.E.; Kleiner-Fisman, G. Characterization of Movement Disorder Phenomenology in Genetically Proven, Familial Frontotemporal Lobar Degeneration: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0153852.
- Xu, X.; Su, Y.; Zou, Z.; Zhou, Y.; Yan, J. Correlation between C9ORF72 mutation and neurodegenerative diseases: A comprehensive review of the literature. Int. J. Med. Sci. 2021, 18, 378–386.
- Estevez-Fraga, C.; Magrinelli, F.; Hensman Moss, D.; Mulroy, E.; Di Lazzaro, G.; Latorre, A.; Mackenzie, M.; Houlden, H.; Tabrizi, S.J.; Bhatia, K.P. Expanding the Spectrum of Movement Disorders Associated With C9orf72 Hexanucleotide Expansions. Neurol. Genet. 2021, 7, e575.
- Benussi, A.; Padovani, A.; Borroni, B. Phenotypic Heterogeneity of Monogenic Frontotemporal Dementia. Front. Aging Neurosci. 2015, 7, 171.
- Mackenzie, I.R. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007, 114, 49–54.
- Kelley, B.J.; Haidar, W.; Boeve, B.F.; Baker, M.; Graff-Radford, N.R.; Krefft, T.; Frank, A.R.; Jack, C.R., Jr.; Shiung, M.; Knopman, D.S.; et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging 2009, 30, 739–751.
- Carecchio, M.; Galimberti, D.; Fenoglio, C.; Serpente, M.; Scarpini, E.; Comi, C.; Terazzi, E.; Cantello, R. Evidence of pre-synaptic dopaminergic deficit in a patient with a novel progranulin mutation presenting with atypical parkinsonism. J. Alzheimer’s Dis. 2014, 38, 747–752.
- Chen, S.; Zhou, R.L.; Zhang, W.; Che, C.H.; Feng, S.Y.; Huang, H.P.; Liu, C.Y.; Zou, Z.Y. Novel TARDBP missense mutation caused familial amyotrophic lateral sclerosis with frontotemporal dementia and parkinsonism. Neurobiol. Aging 2021, 107, 168–173.
- Baizabal-Carvallo, J.F.; Jankovic, J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 2016, 12, 175–185.
- Siuda, J.; Fujioka, S.; Wszolek, Z.K. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat. Disord. 2014, 20, 957–964.
- Arienti, F.; Lazzeri, G.; Vizziello, M.; Monfrini, E.; Bresolin, N.; Saetti, M.C.; Picillo, M.; Franco, G.; Di Fonzo, A. Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review. Cells 2021, 10, 171.
- Kertesz, A.; McMonagle, P.; Jesso, S. Extrapyramidal syndromes in frontotemporal degeneration. J. Mol. Neurosci. 2011, 45, 336–342.
- Dulski, J.; Cerquera-Cleves, C.; Milanowski, L.; Kidd, A.; Sitek, E.J.; Strongosky, A.; Vanegas Monroy, A.M.; Dickson, D.W.; Ross, O.A.; Pentela-Nowicka, J.; et al. Clinical, pathological and genetic characteristics of Perry disease-new cases and literature review. Eur. J. Neurol. 2021, 28, 4010–4021.
- Milanowski, L.; Sitek, E.J.; Dulski, J.; Cerquera-Cleves, C.; Gomez, J.D.; Brockhuis, B.; Schinwelski, M.; Kluj-Kozlowska, K.; Ross, O.A.; Slawek, J.; et al. Cognitive and behavioral profile of Perry syndrome in two families. Parkinsonism Relat. Disord. 2020, 77, 114–120.
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018, 38, 98–107.
More