Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by James B Ames and Version 2 by Bruce Ren.
L-type voltage-gated Ca2+ channels (CaV1.2 and CaV1.3, called CaV) interact with the Ca2+ sensor proteins, calmodulin (CaM) and Ca2+ binding Protein 1 (CaBP1), that oppositely control Ca2+-dependent channel activity. CaM and CaBP1 can each bind to the IQ-motif within the C-terminal cytosolic domain of CaV, which promotes increased channel open probability under basal conditions. At elevated cytosolic Ca2+ levels (caused by CaV channel opening), Ca2+-bound CaM binding to CaV is essential for promoting rapid Ca2+-dependent channel inactivation (CDI). By contrast, CaV binding to CaBP1 prevents CDI and promotes Ca2+-induced channel opening (called CDF).
- calmodulin
- CaBP1
- CaV1.2
- CaV1.3
- L-type Ca2+ channel
- EF-hand
- IQ-motif
1. Introduction
1.1. Voltage-Gated L-Type Ca2+ Channel Structure and Function
Synaptic transmission and neuronal excitability are regulated by the L-type voltage-gated Ca2+ channels (CaV1.2 and CaV1.3, called CaV) expressed in the brain and heart [1][2][3][4][1,2,3,4]. CaVs display slow voltage-dependent gating characteristics (L-type) and are sensitive to a number of different dihydropyridine (DHP) antagonists and agonists [5]. Under resting basal conditions, intracellular Ca2+ concentration is kept low (100 nM) due to the powerful action of Ca2+ pumps and exchangers [1][6][1,6] and Ca2+ sequestration into stores [1][7][1,7]. The opening of CaV channels causes intracellular Ca2+ levels to increase into the micromolar range [8]. This Ca2+ influx triggers a wide range of Ca2+-dependent processes including gene transcription [9], neurotransmitter release [10], neurite outgrowth [11], and the activation of Ca2+-dependent enzymes [12]. Prolonged elevation of intracellular Ca2+ levels is cytotoxic [13], and CaV channels are negatively regulated by a process known as Ca2+-dependent inactivation (CDI) [14][15][16][14,15,16]. Dysregulation of CaVs are linked to various types of neurological disorders, including epilepsy, migraine, and chronic pain [17].
The CaVs are a heteromultimeric protein complex formed by a pore-forming α-subunit and regulatory β and δ subunits (Figure 1). The α-subunit contains four major transmembrane domains (Figure 1A), each with six membrane-spanning helices (termed S1–S6) and a positively charged S4 segment that controls voltage-dependent activation [18]. The transmembrane domains are connected by long cytoplasmic linkers (III-IV inactivation gate [19]), bracketed by cytoplasmic N-terminal and C-terminal domains [20]. The C-terminal domain (residues 1508-1665, called CT1) is important for Ca2+-dependent regulation of channel function and contains important sites (EF-hand and IQ motifs) for protein–protein interactions [21][22][23][21,22,23]. A three-dimensional structure of the skeletal muscle CaV (called CaV1.1) in the absence of CaM was solved by cryo-EM (Figure 1C) [24][25][24,25]. The CaV1.1 structure reveals long-range contacts between the inactivation gate (III-IV linker) and the channel EF-hand domain (orange in Figure 1C), which may undergo Ca2+-induced conformational changes during CDI (see Section 3 below).
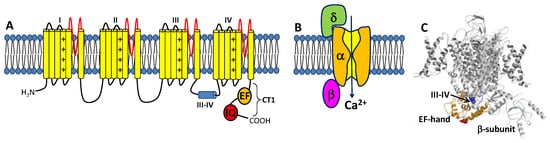
Figure 1. Structure of CaVs. (A) The α-subunit consists of 4 transmembrane domains (I-IV) that contain 6 helices (yellow) and pore loop (red). The III-IV linker is the inactivation gate. The cytosolic C-terminal domain (CT1) is comprised of an EF-hand domain (orange) and IQ-motif (red). (B) CaV is composed of pore-forming α-subunit attached to β- and δ-subunits. (C) Cryo-EM structure of CaV1.1 (PDB ID: 5GJW) showing the inactivation gate (III-IV linker in blue) connected to the EF-hand domain (orange). The IQ-motif is structurally disordered and missing in the cryo-EM structure of CaV1.1.
CaV channels inactivate rapidly by a process known as CDI (Figure 2) that depends critically on CaM [16][26][16,26] and CaBP1 [27][28][27,28]. Ca2+-free CaM is believed to be pre-associated with the CT1 domain such that the C-lobe of CaM interacts with the ‘‘IQ’’ domain and the N-lobe may interact with the EF-hand in order to increase the channel open probability under basal conditions [29][30][31][29,30,31]. Membrane depolarization causes CaV channel opening, which promotes a rise in intracellular Ca2+ that causes a conformational change in the CaV/CaM complex and gives rise to rapid channel inactivation called CDI [29][32][33][34][29,32,33,34]. CaBP1 competes with CaM for binding to CT1 [2][35][2,35], which prevents channel pre-association of CaM and abolishes CDI (Figure 2B).
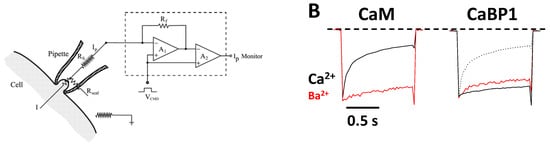
Figure 2. Ca2+-dependent Inactivation (CDI) of CaV. (A) Schematic representation of the electrophysiology experiment used to record CDI. (B) Normalized Ca2+ and Ba2+ currents evoked by 1 s pulse (−80 to +10 mV). Adapted from [2]. Fast decay of Ca2+ current due to CaM (black trace in left panel, CDI). The decay of the Ca2+ current is much slower in the presence of CaBP1 (black solid trace in the right panel, CDI abolished). Dotted line is the Ca2+ current in the absence of CaBP1, caused by endogenous CaM. Red traces are Ba2+ currents that lack fast inactivation because Ba2+ does not bind to CaM.
1.2. CaM Is a Ca2+ Sensor for CaVs
CaM is a 16.7 kDa Ca2+ sensor protein that belongs to the EF-hand superfamily [36]. CaM contains four EF-hand motifs (EF1, EF2, EF3, and EF4) that are grouped into two domains that are separately folded (EF1 and EF2 form the CaM N-lobe, while EF3 and EF4 form the CaM C-lobe) [37]. The CaM C-lobe and N-lobe each bind to Ca2+ with a dissociation constant of ~1 μM and 10 μM, respectively [38]. Thus, Ca2+ binding to CaM is an ordered process in which two Ca2+ bind to the C-lobe first before binding to the N-lobe. The Ca2+-bound form of CaM is known to bind to hundreds of different target proteins, including dozens of enzymes, receptors, ion channels, and other Ca2+ transporters [39]. The Ca2+-induced binding of CaM to its various target proteins usually serves to augment the biological activity of the target protein.
The binding of CaM to CaVs is critically important for promoting CDI [16][26][16,26]. In particular, CaM has been shown to bind to the IQ-motif (residues 1640–1665, highlighted red in Figure 1A) within the C-terminal cytosolic domain of CaVs [40], because deletion of the IQ-motif prevents CaV binding to CaM [26]. The NMR structure of Ca2+-free CaM (apoCaM) bound to the IQ-motif reveals that the IQ peptide forms an α-helix that interacts solely with the CaM C-lobe, while the IQ helix does not interact with the apoCaM N-lobe (Figure 3A). The most prominent intermolecular contacts involve IQ residues I1654 and K1662, and the mutations I1654E and K1662E each weaken apoCaM binding by nearly 10-fold [41]. The crystal structure of Ca2+-bound CaM bound to the IQ-motif reveals that both CaM lobes bind to opposite sides of the IQ helix (Figure 3B). The CaM C-lobe forms hydrophobic contacts with IQ residues I1654 and Q1655 that are essential for binding [42], hence the name IQ-motif. The CaM N-lobe forms hydrophobic contacts with aromatic IQ residues (Y1649 and F1652) that are essential for N-lobe binding. CaV mutations in the IQ-motif (I1654E and I1654M) that weaken CaM binding abolish CDI [43]. Much is known about how CaM interacts with the IQ-motif, but less is known about how the CaM-IQ interaction leads to channel inactivation. In this review, I present the possible molecular mechanisms of CDI to suggest how conformational changes in CaM and CaV might lead to CDI.
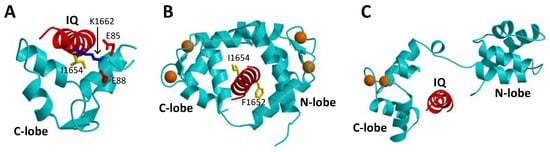
Figure 3. Atomic-level structures of CaM and CaBP1. (A) NMR structure of Ca2+-free CaM C-lobe (cyan) bound to the CaV1.2 IQ-motif in red (PDB ID: 6CTB) [41]. (B) Crystal structure of Ca2+-bound CaM (cyan) bound to the CaV1.2 IQ-motif in red (PDB ID: 2BE6) [44]. (C) Structural model of the crystal structure of CaBP1 (PDB ID: 3OX6) [45] bound to the CaV1.2 IQ-motif (red). Bound Ca2+ are indicated by orange spheres.
1.3. CaBP1 Promotes Activation of CaVs
Neuronal Ca2+-binding proteins (CaBP1-5 [46]) represent a sub-branch of the CaM superfamily [39] that regulate various Ca2+ channel targets. Multiple splice-variants and isoforms of CaBPs are localized in different neuronal cell types [47][48][49][47,48,49] and perform specialized roles in signal transduction. CaBP1, also termed caldendrin [50], has been shown to modulate the Ca2+-sensitive activity of L-type channels [51], and the transient receptor potential channel, TRPC5 [52]. CaBP1 contains four EF-hands, similar in sequence to those found in CaM [39]. By analogy to CaM [37], the four EF-hands are grouped into two domains connected by a central linker that is four residues longer in CaBP1 than in CaM. In contrast to CaM, the first and second EF-hands of CaBP1 lack critical residues required for high affinity Ca2+ binding [46]. CaBP1 binds Ca2+ only at EF3 and EF4, whereas it binds Mg2+ at EF1 that may serve a functional role [53]. In addition to binding Ca2+, CaBP1 also binds tightly to the CaV IQ-motif [35]. A crystal structure is known for CaBP1 with Ca2+ bound to EF3 and EF4 (Figure 3C) [45]. A structural model of CaBP1 bound to the IQ-motif (Figure 3C) was generated here by homology modeling that was calculated based on the crystal structure of the CaM-IQ complex [44]. In this model, the Ca2+-bound CaBP1 C-lobe makes hydrophobic intermolecular contacts with IQ residues I1654 and Y1657, whereas the CaBP1 N-lobe does not make any intermolecular contacts. Future structural and mutagenesis studies of CaBP1 bound to the IQ-motif are needed to test the validity of the structural model in Figure 3C.
The binding of CaBP1 to CaV has been shown to increase the channel open probability and to abolish or prevent CDI. Unlike CaM, CaBP1 appears to cause CaV channel activation at high cytosolic Ca2+ levels, which gives rise to CaV channel CDF [45]. CaBP1 has been suggested to bind to multiple sites within CaV [54]; however, CaBP1 binding to the IQ-motif is believed to cause CDF [55]. The CaBP1 binding to the IQ-motif under basal conditions [35] may serve to block CaM binding to CaV, which may explain how CaBP1 prevents CDI. Schematic mechanisms are presented below to speculate how CaBP1 binding to CaV might activate channel open probability and prevent CDI.
2. CaV Channel Function Regulated by CaM and CaBP1
2.1. CaM Is Both an Accelerator and a Brake for CaV Channel Activity
Neuronal excitability is modulated in part by the Ca2+-dependent activity of CaV channels localized at the synaptic membrane. CaM binding to CaV serves to increase channel activity at low cytosolic Ca2+ levels under basal conditions ([Ca2+]i = 100 nM). Conversely, CaM decreases CaV channel activity at higher cytosolic Ca2+ levels (([Ca2+]i = 1.0 μM) caused by neuronal stimulation. Thus, CaM acts as both an accelerator and a brake to control CaV channel opening [16]. Ca2+ influx through CaV channels causes elevated intracellular Ca2+ levels that in turn promote a rapid negative feedback channel inactivation (called Ca2+-dependent inactivation or CDI [16]), mediated by CaM (Figure 4). Rapid CDI requires CaM to be pre-associated with CaV under basal conditions [29][33][29,33]. The channel has been suggested to be pre-associated with apoCaM under basal conditions (Figure 4A) [35], and apoCaM binding to CaV may increase Ca2+ currents (ICa) and channel open probability (Po) [56], whereas ICa is dramatically decreased at elevated Ca2+ levels, because Ca2+-bound CaM inactivates the channel [15][16][15,16]. As a result, apoCaM binding to CaV in which the CaM C-lobe is bound to the IQ motif (red box in Figure 4) and CaM N-lobe is bound to the channel EF-hand (orange box in Figure 4) is believed to stabilize the channel in the open state at low Ca2+ levels under basal conditions (Figure 4B). At elevated Ca2+ levels (caused by neuronal stimulation), Ca2+-saturated CaM has been suggested to bind to the full-length CaV at two different sites: The N-lobe binds to the NSCaTE domain [15][57][15,57] and the CaM C-lobe binds to the IQ motif [44], which is hypothesized to stabilize the channel in the inactive state (Figure 4C). Atomic-level structures are known for Ca2+/CaM bound to IQ [44] and NSCaTE [57] domains. However, structures are not yet known for apoCaM and Ca2+/CaM each bound to the entire C-terminal cytosolic domain of CaV comprised of the channel EF-hand and IQ-motif (called CT1 domain, Figure 4C). Future studies are needed to elucidate the structural interaction of apoCaM and Ca2+/CaM each bound to the full-length channel to further test the model in Figure 4.
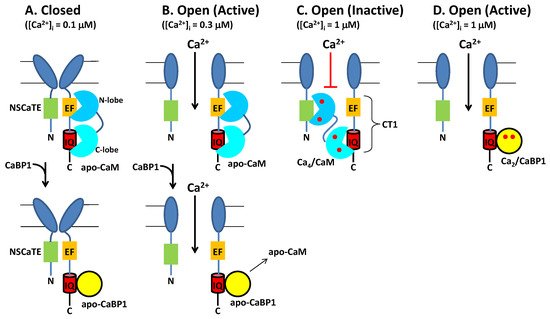
Figure 4. Conventional Model of CDI from CaV regulated by CaM and CaBP1. (A) Under resting conditions ([Ca2+]i = 100 nM), CaV (dark blue) is in the closed channel state, which has been suggested to be pre-associated with Ca2+-free forms of CaM (cyan) or CaBP1 (yellow). (B) Membrane depolarization causes channel opening, which causes Ca2+ influx. Initially at low cytosolic Ca2+ levels (([Ca2+]i < 300 nM), CaV is bound to Ca2+-free forms of CaM or CaBP1, which stabilize the active open state. (C) After sufficient Ca2+ influx, the cytosolic Ca2+ level increases to above 1 micromolar, which causes Ca2+ binding to CaM and the Ca2+-bound CaM promotes channel inactivation (CDI). Alternatively, CaV binding to CaBP1 (yellow) displaces CaM and prevents CDI (bottom panel). (D) The binding of Ca2+-bound CaBP1 to CaV promotes channel opening at elevated Ca2+ levels (called CDF). Bound Ca2+ are indicated by red circles.
2.2. CaBP1 Binding to CaV Prevents CDI and Activates Channel Opening
The upregulated expression of excess CaBP1 in particular neuronal cell types is known to abolish CDI of CaV [2][28][35][51][2,28,35,51] (Figure 4B, lower panel). The Ca2+-bound CaBP1 also increases CaV channel activity (Figure 4D) during Timothy Syndrome [58] and CaBP1 binding to CaVs could be targeted by therapeutics for the disease. CaBP1 was shown to compete with CaM for binding to the IQ-motif [35]. Thus, excess CaBP1 binds to the IQ motif and displaces apoCaM by mass action at low basal Ca2+ levels to prevent CaM-mediated CDI (Figure 4B, bottom panel). Previous studies have suggested that CaBP1 may bind to additional sites within CaV [54]. However, CaBP1 binding to the IQ-motif alone (as depicted in Figure 4) is believed to cause increased NPo under basal conditions and suppress CDI [28][55][28,55]. Future studies are needed to elucidate the atomic-level structural interactions between CaBP1 and CaV to further test the model in Figure 4.