Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yong-Chen Lu and Version 2 by Camila Xu.
Neoantigens are mutated proteins that arise from tumor-specific mutations.
- immunotherapy
- neoantigen
- T cell
- melanoma
1. Neoantigens
Neoantigens are mutated proteins that arise from tumor-specific mutations. The current evidence supports the hypothesis that neoantigens may provide opportunities for T cells to recognize tumor cells [1][2][55,56]. Figure 1 summarizes the interaction between a neoantigen-reactive T cell and a tumor cell. Neoantigens may be generated from alternations in the genomic DNA, or in the post-transcriptional or post-translational steps. These neoantigens are then processed and presented on the tumor cell surface, after which the neoantigens may be recognized by a neoantigen-reactive T cell. Alternatively, an antigen-presenting cell may take neoantigens which were originally generated from a tumor cell and then present them to a neoantigen-reactive T cell. In the following sections, we will describe the sources of neoantigens and the approaches to the identification of these neoantigens.
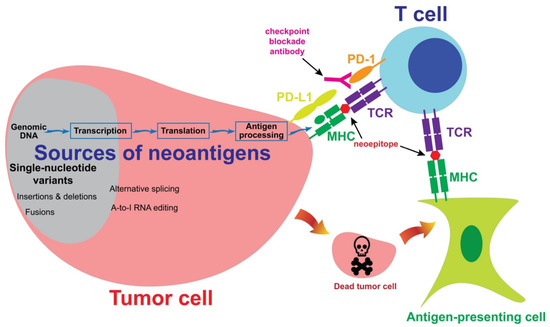
Figure 1. The sources of neoantigens. In cutaneous melanoma, the majority of neoantigens are generated from single-nucleotide variants. Other sources of neoantigens include insertions, deletions, fusions and alternative splicing. A subset of T cells may recognize neoepitopes presented by tumor cells or antigen-presenting cells. Neoantigen-reactive T cells then kill tumor cells in an antigen-specific manner. Co-inhibitory molecules, such as PD-1, may inhibit T cell-mediated immune responses against tumor cells. A checkpoint blockade antibody may block the inhibitory signaling and enhance immune responses.
2. Sources of Neoantigens
2.1. Single-Nucleotide Variants
The most dominant source of neoantigens in melanoma comes from single-nucleotide variants (Figure 1). The majority of melanomas are caused by UV light-induced DNA damage, which leads to cytidine to thymidine (C > T) transitions in genomic DNA [3][4][57,58]. After transcription and translation, some of these C > T mutations produce amino acid changes, also known as nonsynonymous mutations. One of the most interesting features of neoantigens generated from single-nucleotide variants is that a T-cell receptor (TCR) may differentiate a single amino acid change and react to the mutated epitope, but not the wild-type epitope. In some rare cases, TCRs do not have sufficient specificities, recognizing both wild-type and mutated epitopes. In such cases, these single-nucleotide variants should not be considered neoantigens.
Mutated CDK4 (R24C) and CTNNB1 (S37F) were among the first melanoma neoantigens to be discovered by cDNA library screening [5][6][59,60]. Since then, many neoantigens from single-nucleotide variants have been identified by the cDNA library screening approach, which were summarized previously [7][61]. Thanks to the development of next-generation sequencing, the identification of neoantigens from single-nucleotide variants has become quite efficient. The development of these approaches will be discussed in Section 3.
2.2. Insertion, Deletion and Fusion
In addition to nonsynonymous SNV, other alternations in the genomic DNA, including insertion, deletion and fusion, also lead to the changes in protein sequences, which and are also potentially recognized by T cells [8][62]. Some of the insertions and deletions lead to changes in reading frames, which are called frameshift mutations. Frameshift mutations generate long peptides that differ from normal peptides. In a personalized neoantigen vaccine study, six patients with melanoma received pools of synthetic long peptides derived from neoantigen sequences, including frameshift mutations. Eleven neoantigens generated from frameshift mutations could be recognized by the T cells in patients’ peripheral blood after vaccination [9][63].
Chromosome abnormalities in tumor cells, such as chromosome translocation, may result in a fusion gene which contains exons from part of gene A and part of gene B. A mutated peptide may possibly be generated at the junction site. Neoantigens generated from fusion genes have not been reported in melanoma. However, an interesting report showed that the product of an in-frame DEK–AFF2 fusion could be recognized by T cell populations isolated from the peripheral blood of a patient with head and neck squamous cell carcinoma [10][64]. DEK–AFF2 fusion-reactive T cells were likely responsible for the complete response of the checkpoint inhibitor therapy observed in this patient. Therefore, fusion genes may potentially generate neoantigens.
2.3. Post-Transcriptional and Post-Translational Alternations
Some of the alternations at the post-transcriptional step may generate different protein sequences in tumor cells, but not in normal cells. Very frequently, new proteins which are not expressed in normal cells may be produced by tumor cells through the mechanism of alternative splicing. In a comprehensive study, 8705 tumor specimens from 32 cancer types were analyzed, and about 930 alternative splicing events on average were detected in tumors, but not in normal samples [11][65]. Alternative splicing may potentially serve as an important source for neoantigens [12][66]. Additionally, alternative start codons or reading frames may potentially generate neoantigens as well. A classic example is the NA-17A antigen. NA-17A peptide was generated from an intron region in the N-acetylglucosaminyltransferase V gene, which was likely the result of an alternative start site or an alternative splicing event. NA-17A can be detected in multiple melanoma cell lines, and can be recognized by a T cell clone isolated from melanoma tumor-infiltrating lymphocytes (TILs) [13][14][67,68].
Another major source of post-transcriptional modification is adenosine (A) to inosine (I) RNA editing. ADAR (adenosine deaminase acting on RNA) enzymes may edit A to I at specific nucleotide sites. Inosine may pair with cytidine, just as guanosine pairs with cytidine, resulting in altered proteins at the translational step [15][16][69,70]. These altered proteins may potentially be processed and presented by MHC molecules on the cell surface. Zhang M et al. reported that peptides from edited cyclin I could be presented on the surface of melanoma cells and detected by TILs [17][71].
3. Approaches to the Identification of Neoantigens
3.1. cDNA Library Screening
The classical approach to the identification of neoantigens utilized cDNA library screening. A cDNA library was made from a melanoma cell line. Pools of cDNA were transfected into a cell line, and then the cells would present the potential neoantigens to T cells. Neoantigens that were recognized by the T-cells were considered positive hits, and these were further studied to isolate the cDNA clones encoding the neoantigens. Nonsynonymous SNVs in the CDK4, MUM1 and CTNNB1 genes were among the first neoantigens isolated from melanoma by this approach [5][6][18][59,60,72]. Other neoantigens identified by this approach were summarized previously [7][61]. However, it could take months to identify a neoantigen using the cDNA library screening approach. With the recent advances in technology, this approach has not been actively used in recent years.
3.2. Neoepitope Prediction
The approach of using predicted neoepitope to identify neoantigen was initiated by Matsushita et al. [19][73]. Nonsynonymous mutations in a mouse sarcoma cell line were identified by whole-exome sequencing. Amino acid sequences near the mutation sites were subjected to an in-silico analysis to identify potential epitopes that could strongly bind to the MHC class I molecule, H-2D or H-2K. Mutated Sptbn1 was then identified as a potential tumor-rejection antigen. In a similar approach, nonsynonymous mutations were identified from melanoma specimens resected from three patients who received adoptive cell therapies with TILs [20][74]. TILs were stimulated with pools of predicted neoepitopes based on MHC class I binding affinities, and seven neoantigens from these three melanoma specimens were identified by this approach. These neoantigens were validated by stimulating TILs with neoepitopes presented by HEK293 or the COS-7 cell line, and the activation of neoantigen-reactive T cells was detected by IFN-γ ELISA or an ELISpot assay. Another method to validate neoepitopes was MHC-tetramer staining. TILs were stained with potential neoepitope-tetramer complexes. Positive staining by flow cytometric analysis was considered as a neoantigen-reactive T cell population [21][22][75,76].
This approach relies on the accuracy of prediction algorithms, which have been improved significantly in recent years. The latest effort was conducted by the Tumor Neoantigen Selection Alliance [23][77]. Twenty-eight teams utilized their own bioinformatics pipelines to predict neoepitopes from six specimens, including three from metastatic melanoma and three from non-small cell lung cancer. By combining the strengths in several pipelines, a PRNA (predicted and recognized neoantigen abundance) metric was developed that prioritized several features, including strong MHC binding affinity, high tumor abundance, high MHC binding stability, and peptide recognition. This PRNA metric could filter out 98% of non-immunogenic peptides, with a precision of over 0.70.
The prediction approach has the advantage of analyzing dozens of samples within a few weeks, and it has been widely used to study the association between the number of neoantigens and the outcomes of checkpoint blockade therapy, as discussed in Section 5.2. On the other hand, the prediction is not completely accurate, and there is still room for improvement. The uncertainty of prediction has to be clearly stated, otherwise the usage of “predicted” neoantigens, rather than “validated” neoantigens, might jeopardize the integrity of scientific findings.
3.3. Tandem Minigene and Long-Peptide Libraries
In order to overcome the imperfection of neoepitope prediction algorithms, a tandem minigene approach was developed [24][78]. Nonsynonymous mutations, mostly SNVs, were identified by whole-exome sequencing for tumor specimens. Each minigene contained an identified mutated amino acid, flanked by 12 normal amino acids on both the N- and C-termini. This minigene could cover all of the different possibilities of neoepitopes ranging from eight to 13 amino acids in length. Next, six to 24 minigenes were joined in tandem, and the cDNA encoding a tandem minigene was synthesized and cloned into an expression vector. A tandem minigene library was then transfected into a cell line to present all of the potential neoepitopes/neoantigens to T cells. Mutated KIF2C and POLA2 neoantigens were identified from two melanoma specimens in the initial study. Similarly to the tandem minigene approach, a synthetic long peptide, corresponding to a minigene, could be used to assemble a neoantigen library for screening. These approaches have been used in several studies [25][26][27][79,80,81]. More significantly, this design, with some minor modifications, has been utilized to develop personalized neoantigen vaccines for patients with melanoma [9][28][29][63,82,83].
3.4. HLA Peptidomics
One of the concerns about neoantigen identification is that the predicted neoepitopes may not naturally present on the MHC/HLA molecules. In order to study the peptides presented on HLA molecules, a HLA peptidomics technique may be utilized. In this technique, tumor cells are lysed and HLA-peptide complexes are purified by immunoprecipitation. Lastly, peptides are eluted from the HLA molecules and analyzed by mass spectrometry. Additionally, mutations identified by whole-exome sequencing may be used to assist in the identification of neoepitopes [30][31][84,85]. Using this approach, five neoantigens were identified in three patients with melanoma in a study [30][84]. Additionally, two of the neoantigen-reactive T cell clones found in this study could kill most of the autologous melanoma cells both in vitro and in vivo. This approach may have a strong potential to accurately identify neoantigens in the future.