COVID-19 and RA share similar immune-inflammatory features of disease pathogenesis executed by analogous mechanistic pathways. However, the treatment of RA patients in the COVID-19 setting itself stands as a challenging task. Implementation of individualized clinical surveillance of RA patients considering the disease severity and appropriate risk-benefit study referring to the recommendations of using anti-rheumatic drugs in the COVID-19 setting by different professional rheumatology associations would stand as the optimal therapeutic strategy for effective disease control during the COVID-19 pandemic.
- ACE
- ACE2
- anti-rheumatic drugs
- COVID-19
- cytokine storm
- immune response
- inflammation
- rheumatoid arthritis
- SARS-CoV-2
- therapeutic options
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection that caused coronavirus disease 2019 (COVID-19) usually produces a mild to moderate respiratory disease [1]. However, it occasionally leads to severe alveolar disease resulting in shortening of breath, reduced oxygen saturation in blood, and pulmonary infiltration in the lung that can substantially contribute to pulmonary failure [2]. Age, the severity of infection, and the existence of comorbidities are potential risk factors in COVID-19 patients [1,3][1][3]. Emerging evidence revealed that SARS-CoV-2 develops a specific type of alveolar disease that is clinically different from other acute respiratory syndromes [2]. Immune hyperactivation and cytokine involvement in alveolar structures have been identified as the key contributors to produce severe lung disease in COVID-19 patients. Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by synovial inflammation and hyperactivation of T cells. Several pro-inflammatory cytokines act as contributing factors in developing synovial inflammation in RA. The patterns of cytokine and immune activation in COVID-19 patients seem to resemble the RA case. Interestingly, some common therapeutic strategies including cytokine inhibition have been found to be fruitful against both COVID-19 and RA [2]. Thus, a possibility of pathological crosstalk is inevitable between COVID-19 and RA.
In general, there is a close association between viral infection and arthritis with a wide spectrum of symptoms ranging from arthralgia to arthritis. Earlier reports revealed that individuals infected with hepatitis C and several alphaviruses frequently develop prolonged arthritis; however, Parvovirus B19, Hepatitis B, and Rubella viruses frequently cause self-limited arthritis. In contrast, respiratory viruses, such as corona and influenza viruses more frequently can cause arthralgia and/or myalgia. Approximately 15 and 44% of COVID-19 patients present arthralgia and/or myalgia, respectively, during the infective stage [4]. Emerging evidence hypothesized that SARS-CoV-2 infection can attack musculoskeletal systems through immune-inflammation-dependent mechanisms, which may develop inflammatory arthritis during the infective or post-infective stage [3,4,5][3][4][5]. However, little is known about the manifestations or worsening of RA by this infection. Since musculoskeletal manifestations phenotypically resemble RA, it has been attempted to find out the association between COVID-19 and RA. In this article, we reviewed the pathological crosstalk between COVID-19 and RA. In addition, our understanding of the risk of RA patients in acquiring SARS-CoV-2 infection and worsening COVID-19 outcomes was critically discussed on the basis of available clinical readouts. The therapeutic strategies and guidelines were conferred referring to recently published literature. Moreover, critical arguments on therapeutic challenges raised in different case studies were discussed in this review.
2. Mechanistic Similarity between SARS-COV-2 Infection and RA
2.1. Angiotensin-Converting Enzyme (ACE)-Dependent Pathway
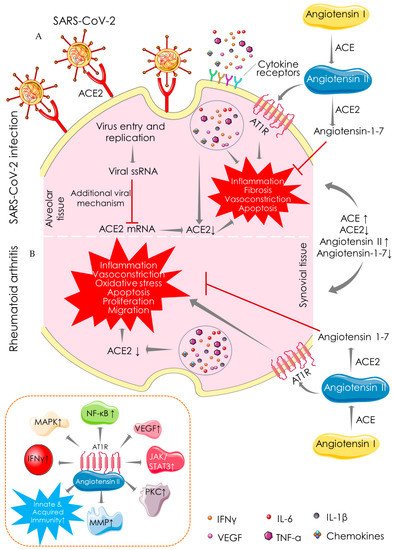
2.2. Macrophage-Mediated Pathway
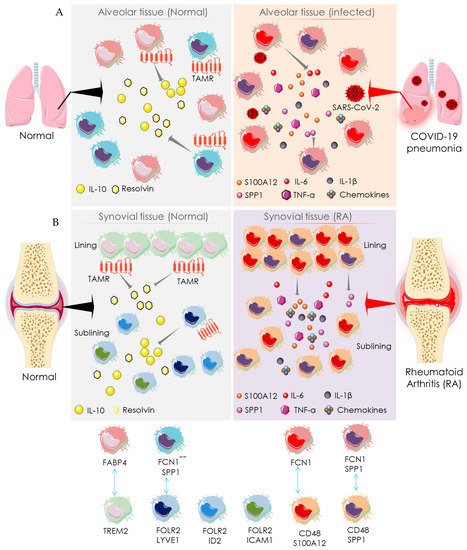
3. Recommendation for Anti-Rheumatic Drugs in the COVID-19 Setting
References
- Kalra, R.S.; Tomar, D.; Meena, A.S.; Kandimalla, R. SARS-CoV-2, ACE2, and Hydroxychloroquine: Cardiovascular Complications, Therapeutics, and Clinical Readouts in the Current Settings. Pathogens 2020, 9, 546.
- Schett, G.; Manger, B.; Simon, D.; Caporali, R. COVID-19 revisiting inflammatory pathways of arthritis. Nat. Rev. Rheumatol. 2020, 16, 465–470.
- Kalra, R.S.; Dhanjal, J.K.; Meena, A.S.; Kalel, V.C.; Dahiya, S.; Singh, B.; Dewanjee, S.; Kandimalla, R. COVID-19, Neuropathology, and Aging: SARS-CoV-2 Neurological Infection, Mechanism, and Associated Complications. Front. Aging Neurosci. 2021, 13, 662786.
- Mukarram, M.S.; Ishaq Ghauri, M.; Sethar, S.; Afsar, N.; Riaz, A.; Ishaq, K. COVID-19: An Emerging Culprit of Inflammatory Arthritis. Case Rep. Rheumatol. 2021, 2021, 6610340.
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Puvvada, N.; Kandimalla, R.; Reddy, P.H. Emerging COVID-19 Neurological Manifestations: Present Outlook and Potential Neurological Challenges in COVID-19 Pandemic. Mol. Neurobiol. 2021, 58, 4694–4715.
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Gao, G. The potential role of serum angiotensin-converting enzyme in coronavirus disease 2019. BMC Infect. Dis. 2020, 20, 883.
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422.
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.
- Choudhary, S.; Sharma, K.; Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb. Pathog. 2021, 150, 104673.
- Kalra, R.S.; Kandimalla, R. Engaging the spikes: Heparan sulfate facilitates SARS-CoV-2 spike protein binding to ACE2 and potentiates viral infection. Signal. Transduct. Target. Ther. 2021, 6, 39.
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Ronconi, G. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: A promising inhibitory strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 1971–1975.
- Rodríguez-Puertas, R. ACE2 activators for the treatment of COVID 19 patients. J. Med. Virol. 2020, 92, 1701–1702.
- Wong, M.C.S.; Wong, S.; Huang, J.; Yan, B. Relating angiotensin-converting enzyme inhibitors or angiotensin receptor blockers with incidence or mortality of COVID-19. ESC Heart Fail. 2020, 7, 3119–3123.
- Chang, Y.; Wei, W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin. Exp. Immunol. 2015, 179, 137–145.
- Najafi, S.; Rajaei, E.; Moallemian, R.; Nokhostin, F. The potential similarities of COVID-19 and autoimmune disease pathogenesis and therapeutic options: New insights approach. Clin. Rheumatol. 2020, 39, 3223–3235.
- Mirastschijski, U.; Dembinski, R.; Maedler, K. Lung Surfactant for Pulmonary Barrier Restoration in Patients with COVID-19 Pneumonia. Front. Med. 2020, 7, 254.
- MacDonald, L.; Alivernini, S.; Tolusso, B.; Elmesmari, A.; Somma, D.; Perniola, S.; Paglionico, A.; Petricca, L.; Bosello, S.L.; Carfì, A.; et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight 2021, 6, e147413.
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306.
- Favalli, E.G.; Maioli, G.; Biggioggero, M.; Caporali, R. Clinical management of patients with rheumatoid arthritis during the COVID-19 pandemic. Expert Rev. Clin. Immunol. 2021, 17, 561–571.
- Akiyama, S.; Hamdeh, S.; Micic, D.; Sakuraba, A. Prevalence and clinical outcomes of COVID-19 in patients with autoimmune diseases: A systematic review and meta-analysis. Ann. Rheum. Dis. 2020, 80, 384–391.
- Roongta, R.; Ghosh, A. Managing rheumatoid arthritis during COVID-19. Clin. Rheumatol. 2020, 39, 3237–3244.
- Dougados, M.; Soubrier, M.; Antunez, A.; Balint, P.; Balsa, A.; Buch, M.H.; Casado, G.; Detert, J.; El-Zorkany, B.; Emery, P.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: Results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 2014, 73, 62–68.
- Mikuls, T.R.; Johnson, S.R.; Fraenkel, L.; Arasaratnam, R.J.; Baden, L.R.; Bermas, B.L.; Chatham, W.; Cohen, S.; Costenbader, K.; Gravallese, E.M.; et al. American College of Rheumatology Guidance for the Management of Rheumatic Disease in Adult Patients During the COVID-19 Pandemic: Version 1. Arthritis Rheumatol. 2020, 72, 1241–1251.
- Landewé, R.B.; Machado, P.M.; Kroon, F.; Bijlsma, H.W.; Burmester, G.R.; Carmona, L.; Combe, B.; Galli, M.; Gossec, L.; Iagnocco, A.; et al. EULAR provisional recommendations for the management of rheumatic and musculoskeletal diseases in the context of SARS-CoV-2. Ann. Rheum. Dis. 2020, 79, 851–858.