Advances in the treatment of pancreatic ductal adenocarcinoma (PDAC) using neoadjuvant chemoradiotherapy, chemotherapy, and immunotherapy have had minimal impact on the overall survival of patients. A general lack of immunogenic features and a complex tumor microenvironment (TME) are likely culprits for therapy refractoriness in PDAC. Induced pluripotent stem cells (iPSCs) should be explored as a means to advance the treatment options for PDAC, by providing representative in vitro models of pancreatic cancer development. In addition, iPSCs could be used for tailor-made cellular immunotherapies or as a source of tumor-associated antigens in the context of vaccination.
- pancreatic ductal adenocarcinoma
- PDAC
- induced pluripotent stem cells
- iPSCs
- cancer models
- cancer vaccine
- cancer therapy
- immunotherapy
- microenvironment
1. Introduction
2. The Potential of Induced Pluripotent Stem Cells (iPSCs)
Pros + | Cons − | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Models | + | Insight into disease onset and development | − | Risk of transcriptional and genetic alterations during iPSC induction | ||||||||||||||
+ | Reliable cell source for multicellular models | − | Heterogeneity within iPSC populations | |||||||||||||||
Immunotherapeutic approaches |
[38][39][40][41][42][43][44][45][46] | (ClincalTrials.gov, Identifier: NCT03841110 | jrct.niph.go.jp, Trial ID: jRCT2033200116) | + | iPSCs are immunogenic and express tumor-associated antigens | − | Oncogenic potential of iPSCs reprogramming factors | |||||||||||
+ | Reliable cell source for off-the-shelf cellular therapies | − | Risk of transcriptional and genetic alterations during iPSC induction | |||||||||||||||
+ | Large-scale cell production | − | Heterogeneity within iPSC populations | |||||||||||||||
3. iPSC-Based PDAC Models and Their Potential for Disease Modeling
4. iPSCs as a Cell-Based Immunotherapy
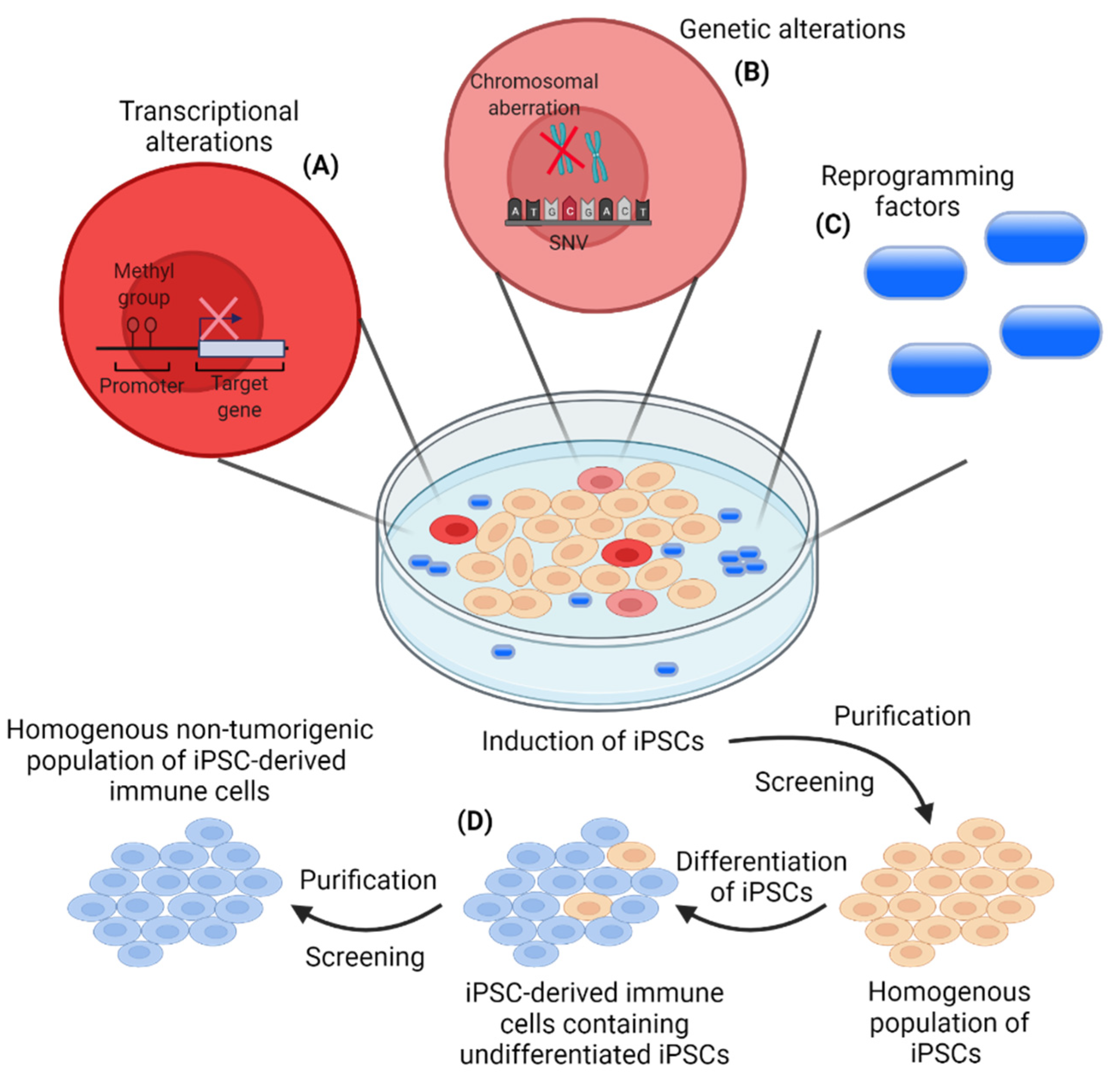
5. iPSC-Derived Immune Cells for Cancer Immunotherapies
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. Cancer Research for Cancer Prevention World Cancer Report; World Health Organization: Lyon, France, 2020.
- National Cancer Institute. SEER Cancer Stat Facts: Pancreatic Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 1 July 2021).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33.
- Hruban, R.H.; Adsay, N.V.; Albores–Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Klöppel, G.; Longnecker, D.S.; et al. Pancreatic Intraepithelial Neoplasia. Am. J. Surg. Pathol. 2001, 25, 579–586.
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86.
- Kopp, J.L.; Von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750.
- Wagner, M.; Lührs, H.; Klöppel, G.; Adler, G.; Schmid, R.M. Malignant transformation of duct-like cells originating from acini in transforming growth factor α transgenic mice. Gastroenterology 1998, 115, 1254–1262.
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047–2060.
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638.
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nat. Cell Biol. 1981, 292, 154–156.
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676.
- González, F.; Boue, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242.
- Malik, N.; Rao, M.S. A Review of the Methods for Human iPSC Derivation. Methods Mol. Biol. 2013, 997, 23–33.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872.
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. science 2007, 318, 1917–1920.
- Shao, L.; Feng, W.; Sun, Y.; Bai, H.; Liu, J.; Currie, C.; Kim, J.; Gama, R.; Wang, Z.; Qian, Z.; et al. Generation of iPS cells using defined factors linked via the self-cleaving 2A sequences in a single open reading frame. Cell Res. 2009, 19, 296–306.
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362.
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of Defective and Persistent Sendai Virus Vector. J. Biol. Chem. 2011, 286, 4760–4771.
- Woltjen, K.; Michael, I.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nat. Cell Biol. 2009, 458, 766–770.
- Anokye-Danso, F.; Trivedi, C.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly Efficient miRNA-Mediated Reprogramming of Mouse and Human Somatic Cells to Pluripotency. Cell Stem Cell 2011, 8, 376–388.
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630.
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476.
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 381–384.
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412.
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells Without Viral Vectors. science 2008, 322, 949–953.
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.; Amoroso, M.W.; Oakley, D.; et al. Reference Maps of Human ES and iPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 2011, 144, 439–452.
- Mallon, B.S.; Hamilton, R.S.; Kozhich, O.A.; Johnson, K.R.; Fann, Y.C.; Rao, M.S.; Robey, P. Comparison of the molecular profiles of human embryonic and induced pluripotent stem cells of isogenic origin. Stem Cell Res. 2014, 12, 376–386.
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.; Fann, Y.C.; et al. StemCellDB: The Human Pluripotent Stem Cell Database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66.
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181.
- Breunig, M.; Merkle, J.; Wagner, M.; Melzer, M.K.; Barth, T.F.; Engleitner, T.; Krumm, J.; Wiedenmann, S.; Cohrs, C.M.; Perkhofer, L.; et al. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell 2021, 28, 1105–1124.
- Calle, A.S.; Nair, N.; Oo, A.K.K.; Prieto-Vila, M.; Koga, M.; Khayrani, A.C.; Seno, M. A New PDAC Mouse Model Originated from iPSCs-Converted Pancreatic Cancer Stem Cells (CSCcm). Am. J. Cancer Res. 2016, 6, 2799–2815.
- Huang, L.; Desai, R.; Conrad, D.N.; Leite, N.C.; Akshinthala, D.; Lim, C.M.; Gonzalez, R.; Muthuswamy, L.B.; Gartner, Z.; Muthuswamy, S.K. Commitment and oncogene-induced plasticity of human stem cell-derived pancreatic acinar and ductal organoids. Cell Stem Cell 2021, 28, 1090–1104.
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.-X.; Won, K.J.; et al. An iPSC Line from Human Pancreatic Ductal Adenocarcinoma Undergoes Early to Invasive Stages of Pancreatic Cancer Progression. Cell Rep. 2013, 3, 2088–2099.
- Hassan, G.; Afify, S.M.; Nair, N.; Kumon, K.; Osman, A.; Du, J.; Mansour, H.; A Abu Quora, H.; Nawara, H.M.; Satoh, A.; et al. Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells. Cancers 2019, 12, 82.
- Matsuda, S.; Yan, T.; Mizutani, A.; Sota, T.; Hiramoto, Y.; Prieto-Vila, M.; Chen, L.; Satoh, A.; Kudoh, T.; Kasai, T.; et al. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int. J. Cancer 2014, 135, 27–36.
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 1–13.
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12, 1–15.
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258.
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953.
- Kooreman, N.G.; Kim, Y.; de Almeida, P.E.; Termglinchan, V.; Diecke, S.; Shao, N.-Y.; Wei, T.-T.; Yi, H.; Dey, D.; Nelakanti, R.; et al. Autologous iPSC-Based Vaccines Elicit Anti-tumor Responses In Vivo. Cell Stem Cell 2018, 22, 501–513.
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5.
- Lu, S.; Zhang, Z.; Du, P.; Chard, L.S.; Yan, W.; El Khouri, M.; Wang, Z.; Zhang, Z.; Chu, Y.; Gao, D.; et al. A Virus-Infected, Reprogrammed Somatic Cell–Derived Tumor Cell (VIReST) Vaccination Regime Can Prevent Initiation and Progression of Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 465–476.
- Ouyang, X.; Liu, Y.; Zhou, Y.; Guo, J.; Wei, T.-T.; Liu, C.; Lee, B.; Chen, B.; Zhang, A.; Casey, K.M.; et al. Antitumor effects of iPSC-based cancer vaccine in pancreatic cancer. Stem Cell Rep. 2021, 16, 1468–1477.
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933.
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 1–5.
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An In vivo Platform for Translational Drug Development in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 4652–4661.
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; E Gallick, G. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323.
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.; et al. Tumor-Initiating Cells Are Rare in Many Human Tumors. Cell Stem Cell 2010, 7, 279–282.
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037.
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A Model of Cancer Stem Cells Derived from Mouse Induced Pluripotent Stem Cells. PLoS ONE 2012, 7, e33544.
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nat. Cell Biol. 2016, 531, 47–52.
- Biankin, A.V.; Initiative, A.P.C.G.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nat. Cell Biol. 2012, 491, 399–405.
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.C.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.C.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, e6744.
- Tu, Q.; Hao, J.; Zhou, X.; Yan, L.; Dai, H.; Sun, B.; Yang, D.; An, S.; Lv, L.; Jiao, B.; et al. CDKN2B deletion is essential for pancreatic cancer development instead of unmeaningful co-deletion due to juxtaposition to CDKN2A. Oncogene 2017, 37, 128–138.
- Waddell, N.; Initiative, A.P.C.G.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nat. Cell Biol. 2015, 518, 495–501.
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, M.C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell– and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371.
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Müller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2016, 66, 473–486.
- Ferreira, R.; Sancho, R.; Messal, H.A.; Nye, E.; Spencer-Dene, B.; Stone, R.K.; Stamp, G.; Rosewell, I.; Quaglia, A.; Behrens, A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017, 21, 966–978.
- Zheng, X.; Carstens, J.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nat. Cell Biol. 2015, 527, 525–530.
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828.
- Rhim, A.D.; Mirek, E.T.; Aiello, N.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361.
- Mitchem, J.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141.
- Lee, C.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725.
- Gutbier, S.; Wanke, F.; Dahm, N.; Rümmelin, A.; Zimmermann, S.; Christensen, K.; Köchl, F.; Rautanen, A.; Hatje, K.; Geering, B.; et al. Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening. Int. J. Mol. Sci. 2020, 21, 4808.
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 1–8.
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, N.M.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778.
- Sunami, Y.; Böker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697.
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505.
- Prieto-Vila, M.; Yan, T.; Calle, A.S.; Nair, N.; Hurley, L.; Kasai, T.; Seno, M. iPSC-Derived Cancer Stem Cells Provide a Model of Tumor Vasculature. Am. J. Cancer Res. 2016, 6, 1906–1921.
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501.
- Brewer, B.G.; Mitchell, R.A.; Harandi, A.; Eaton, J.W. Embryonic vaccines against cancer: An early history. Exp. Mol. Pathol. 2009, 86, 192–197.
- Ghosh, Z.; Huang, M.; Hu, S.; Wilson, K.D.; Dey, D.; Wu, J.C. Dissecting the Oncogenic and Tumorigenic Potential of Differentiated Human Induced Pluripotent Stem Cells and Human Embryonic Stem Cells. Cancer Res. 2011, 71, 5030–5039.
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; A Weinberg, R. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507.
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nat. Cell Biol. 2011, 474, 212–215.
- De Almeida, P.E.; Meyer, E.H.; Kooreman, N.G.; Diecke, S.; Dey, D.; Sanchez-Freire, V.; Hu, S.; Ebert, A.D.; I Odegaard, J.; Mordwinkin, N.M.; et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014, 5, 3903.
- Tabatabai, R.; Linhares, Y.; Bolos, D.; Mita, M.; Mita, A. Targeting the Wnt Pathway in Cancer: A Review of Novel Therapeutics. Target. Oncol. 2017, 12, 623–641.
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942.
- de Jesus, B.B.; Neves, B.M.; Ferreira, M.; Nóbrega-Pereira, S. Strategies for Cancer Immunotherapy Using Induced Pluripoten-cy Stem Cells-Based Vaccines. Cancers 2020, 12, 3581.
- Gąbka-Buszek, A.; Kwiatkowska-Borowczyk, E.; Jankowski, J.; Kozłowska, A.K.; Mackiewicz, A. Novel Genetic Melanoma Vaccines Based on Induced Pluripotent Stem Cells or Melanosphere-Derived Stem-Like Cells Display High Efficacy in a murine Tumor Rejection Model. Vaccines 2020, 8, 147.
- Wang, J.; Shao, L.; Wu, L.; Ma, W.; Zheng, Y.; Hu, C.; Li, F. Expression levels of a gene signature in hiPSC associated with lung adenocarcinoma stem cells and its capability in eliciting specific antitumor immune-response in a humanized mice model. Thorac. Cancer 2020, 11, 1603–1612.
- Li, Y.; Zeng, H.; Xu, R.-H.; Liu, B.; Li, Z. Vaccination with Human Pluripotent Stem Cells Generates a Broad Spectrum of Immunological and Clinical Response against Colon Cancer. Stem Cells 2009, 27, 3103–3111.
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004.
- Kooreman, N.G.; Wu, J.C. Tumorigenicity of pluripotent stem cells: Biological insights from molecular imaging. J. R. Soc. Interface 2010, 7, S753–S763.
- Koyanagi-Aoi, M.; Ohnuki, M.; Takahashi, K.; Okita, K.; Noma, H.; Sawamura, Y.; Teramoto, I.; Narita, M.; Sato, Y.; Ichisaka, T.; et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20569–20574.
- Kawamura, A.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ito, E.; Watabe, T.; Masuda, S.; Toda, K.; Hatazawa, J.; et al. Teratocarcinomas Arising from Allogeneic Induced Pluripotent Stem Cell-Derived Cardiac Tissue Constructs Provoked Host Immune Rejection in Mice. Sci. Rep. 2016, 6, 19464.
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic Expression of Oct-4 Blocks Progenitor-Cell Differentiation and Causes Dysplasia in Epithelial Tissues. Cell 2005, 121, 465–477.
- Zhou, X.; Huang, G.-R.; Hu, P.; Xi, Z.; Guang-Rong, H.; Pin, H. Over-expression of Oct4 in human esophageal squamous cell carcinoma. Mol. Cells 2011, 32, 39–45.
- Riggi, N.; Suvà, M.-L.; De Vito, C.; Provero, P.; Stehle, J.-C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932.
- Zhao, F.-Q.; Misra, Y.; Li, D.-B.; Wadsworth, M.P.; Krag, D.; Weaver, D.; Tessitore, J.; Zhang, G.; Tian, Q.; Buss, K. Differential expression of Oct3/4 in human breast cancer and normal tissues. Int. J. Oncol. 2018, 52, 2069–2078.
- Bae, K.-M.; Su, Z.; Frye, C.; McClellan, S.; Allan, R.W.; Andrejewski, J.T.; Kelley, V.; Jorgensen, M.; Steindler, D.A.; Vieweg, J.; et al. Expression of Pluripotent Stem Cell Reprogramming Factors by Prostate Tumor Initiating Cells. J. Urol. 2010, 183, 2045–2053.
- Sarkar, A.; Hochedlinger, K. The Sox Family of Transcription Factors: Versatile Regulators of Stem and Progenitor Cell Fate. Cell Stem Cell 2013, 12, 15–30.
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35.
- Clemente-Periván, S.I.; Gómez-Gómez, Y.; Leyva-Vázquez, M.A.; Lagunas-Martínez, A.; Organista-Nava, J.; Illades-Aguiar, B. Role of Oct3/4 in Cervical Cancer Tumorigenesis. Front. Oncol. 2020, 10, 247.
- Themeli, M.; Rivière, I.; Sadelain, M. New Cell Sources for T Cell Engineering and Adoptive Immunotherapy. Cell Stem Cell 2015, 16, 357–366.