Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sergio Piña-Oviedo and Version 1 by Sergio Piña-Oviedo.
Anaplastic large cell lymphoma (ALCL) is a subtype of T-cell lymphoma composed of large cells and a characteristic strong and diffuse expression of the activation marker CD30 (>75% of the cells). ALCL is composed of large epithelioid and anaplastic cells and cells with horseshoe-shaped nucleus (hallmark cells). This lymphoma is divided into two major groups based on the presence/absence of the rearrangement and of the expression of anaplastic lymphoma kinase (ALK) into ALK-positive and ALK-negative. The latter is currently classified into systemic, primary cutaneous, and breast implant-associated ALCL.
- Immunohistochemistry
- anaplastic lymphoma kinase (ALK)
- T-cell lymphoma
- anaplastic large cell lymphoma
- pathology
- CD30
1. Background
Anaplastic large cell lymphoma (ALCL) is a subtype of T-cell lymphoma (TCL) composed of large cells and a characteristic strong and diffuse expression of the activation marker CD30. This lymphoma is composed of large epithelioid cells with anaplastic features, namely significant atypia and marked pleomorphism.
ALCL is divided into two major groups based on the presence/absence of the rearrangement and of the expression of the anaplastic lymphoma kinase (ALK) into ALK-positive (ALK+) and ALK-negative (ALK-), with each group roughly comprising 50% of cases [1]. ALK+ ALCL is more common in the pediatric population and has a good overall prognosis (overall survival 70–80%), whereas ALK- ALCL is more common in adults and has a more aggressive behavior (overall survival 40–60%), although prognosis may also be related to the age of presentation rather than to ALK expression [2,3]. In contrast to ALK+ ALCL where the expression of ALK and CD30 virtually excludes any other diagnostic possibility regardless of the morphology, the distinction between ALK- ALCL and other large TCLs with CD30 expression is difficult.
Based on its location and extension of involvement, ALK- ALCL is currently classified into (1) systemic, (2) primary cutaneous (pc-ALCL), and (3) breast implant-associated ALCL (BIA-ALCL) [1]. Recent molecular studies have identified diverse genetic alterations in ALK- ALCL that convey different prognoses and have different underlying pathogenetic mechanisms, obviating the heterogeneity of this group of lymphomas. It is very likely that in the coming years systemic ALK- ALCL may be further divided into molecular subtypes according to the presence/absence of genetic abnormalities.
2. Systemic ALK- ALCL
2.1. Definition
This lymphoma is defined as the involvement of the lymph nodes, bone marrow, and/or extranodal organs by ALK- ALCL at initial presentation. Pc-ALCL and BIA-ALCL are excluded from this group, but it is difficult to distinguish systemic disease from multiorgan spread from these two subtypes and only a prior clinical history of cutaneous or breast-implant associated disease may point to this possibility.
2.2. Historical Aspects
Most cases of ALCL were categorized in the past as non-hematopoietic neoplasms or as tumors of histiocytic or reticulum cell origin [4]. It was not until the application of immunohistochemistry and the capability to grow cultures of Reed-Sternberg cells from classic Hodgkin lymphoma (CHL) by V. Diehl, H. Stein, and cols. in Germany that the discovery of the CD30 antigen was possible in 1982 [5]. This antigen was abundant in Reed-Sternberg cells and received the name “Ki-1” (“Ki” from “Kiel” and “1” alluding to the number of the antibody clone; later designated CD30). In 1985, H. Stein et al. recognized that a group of anaplastic hematopoietic tumors with particular nodal involvement were positive for Ki-1 and frequently positive for ≥1 T-cell markers and designated these neoplasms “ALCL” or “anaplastic Ki-1 LCL” [6]. ALCL was then the preferred name for this neoplasm in the Kiel classification of 1988 as well as in the REAL classification of 1994 [7,8]. In the REAL classification, ALCL was recognized as a tumor of T-cell or a “null” phenotype different from CD30+ large B-cell lymphomas.
Since there could not be an ALK- ALCL without ALK, one could say that the discovery of this lymphoma technically did not occur until 1994 when S. Morris, T. Look and cols. at that time at Saint Jude Children’s Hospital discovered the genes involved in the translocation t(2;5)(p23;q35) and recognized a novel tyrosine kinase in chromosome 2 that was called anaplastic lymphoma kinase (ALK) [9]. ALK- ALCL was included as a provisional entity in the 2008 World Health Organization (WHO) classification and is currently classified as a distinct entity in the current 2017 WHO revised 4th edition [1,10].
2.3. Epidemiology, Risk Factors, and Clinical Features
TCLs represent 10 to 15% of all non-Hodgkin lymphomas [11,12]. ALK+ and ALK- ALCL combined represent ~2% of all adult non-Hodgkin lymphomas and are the fourth most common TCL (~10%) after peripheral TCL, not otherwise specified, mycoses fungoides, and angioimmunoblastic TCL [11,12].
Most individuals affected by ALK- ALCL are adults (40 to 65 years). The male to female ratio is 1.5:1 [13]. They usually present at advanced stages of the disease with systemic symptoms. ALK- ALCL involves the lymph nodes in ~50% of cases whereas extranodal involvement is less frequent [14,15]. Relatively common extranodal sites of involvement include the skin (excluding pc-ALCL), soft tissue, the liver and the lungs [3,16,17,18,19,20,21,22,23]. Skin lesions may be solitary or multiple in the form of papules, nodules, or tumors. Leukemic presentation is very rare [24,25,26,27].
Based on data from the InterLymph Non-Hodgkin Lymphoma Subtypes Project, textile workers and electrical fitters have a significantly increased risk of ALCL (OR of 4.08 and 2.60, respectively) but according to the authors the sample size was modest and further information on ALK status was not sufficient for more definitive conclusions to be made [28]. Similarly, there are not enough data to establish an association between autoimmune disorders or an immunocompromised status and the development of ALK- ALCL [13]. Importantly, BIA-ALCL develops in the setting of long-standing textured breast implants [29,30,31].
ALK- ALCL is not typically associated with infection by the Epstein-Barr virus (EBV). Similarly, no association with infection by the human T-cell leukemia virus type 1 (HTLV-1) or the human immunodeficiency virus (HIV) has been recognized.
2.4. Pathology
A lymph node involved with ALCL is tan-white to pale-pink with variable hemorrhage and necrosis. At extranodal sites it presents as a mass with these same features. As defined by the WHO in 2017, ALK- ALCL is a “CD30+ T-cell neoplasm that is not reproducibly distinguishable on morphologic grounds from ALK+ ALCL, but lacks (ALK) protein expression” [1]. Therefore, the morphologic features described below are seen in both ALK+ and ALK- ALCL (common or classic variant). These tumors are composed of sheets of large epithelioid cells with variable degrees of pleomorphism and anaplasia (Figure 1A,B). The lymphoma cells have an eccentric nucleus with vesicular to moderately condensed chromatin, a prominent nucleolus, and an abundant eosinophilic or amphophilic cytoplasm with a prominent Golgi zone. The characteristic cells of ALCL have a horseshoe-shaped nucleus (“hallmark” cells of Benharroch and Delsol [34]) and are present in all cases in variable amounts (Figure 1A,B). In addition, tumor cells may be binucleated (Reed-Sternberg-like), may have a ring-shaped nucleus with a central pseudoinclusion (“doughnut” cells), may be multinucleated with nuclei arranged in a wreathlike configuration, or may have a markedly convoluted (“embryoid”) nucleus. Even though morphology cannot reliably distinguish between ALK+ and ALK- ALCL, certain features might suggest that a case is ALK-, including the presence of plasmablastic features, and/or the presence of a starry-sky pattern (Figure 1C,D). Similarly, cases of ALK- ALCL harboring DUSP22 rearrangements are more likely to contain “doughnut” cells, less pleomorphism, and a sheet-like growth with an insignificant number of background inflammatory cells (Figure 1C,D) [35]. Nevertheless, ALK immunohistochemistry is mandatory to confirm the diagnosis.
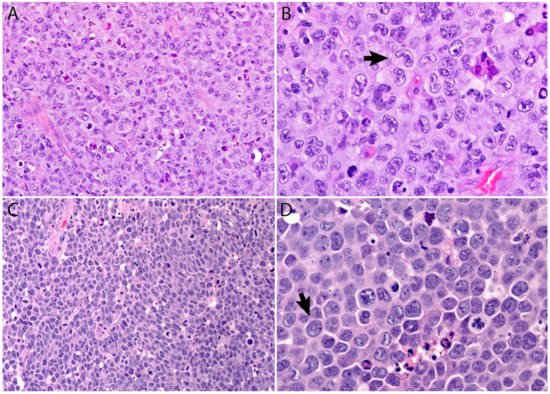
Figure 1. (A,B) ALCL is a neoplasm composed of sheets of large pleomorphic/anaplastic tumor cells with abundant cytoplasm. Occasional multinucleated cells and Reed-Sternberg-like cells are seen. (B) “Hallmark” cells are characteristic of this neoplasm (arrow). (C,D) Most cases of ALK- ALCL with DUSP22 rearrangement have a more monotonous appearance and are less pleomorphic. This case has a starry-sky appearance. (D) Cells with central nuclear pseudoinclusions or a “doughnut” nuclear shape are also seen (arrow).
Additional features include the presence of frequent mitoses and variable necrosis. Nodal involvement demonstrates a characteristic sinusoidal distribution of the tumor cells mimicking metastatic carcinoma, metastatic melanoma, or histiocytic sarcoma (Figure 2A). In cases with partial nodal involvement, the tumor cells are preferentially located in the interfollicular region (Figure 2B).
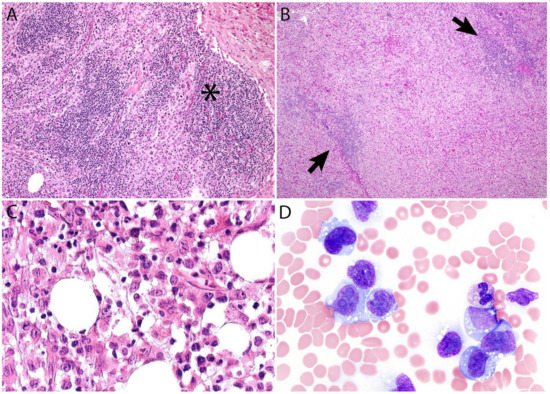
Figure 2. (A) Sinusoidal distribution of ALCL mimicking metastatic carcinoma (asterisk). (B) ALCL with marked expansion of the paracortical area and subtotal effacement of a lymph node. The arrows point to residual compressed lymphoid follicles. (C) ALK- ALCL involving bone marrow. (D) Leukemic ALK- ALCL mimicking monocytic leukemia.
Bone marrow involvement may be readily identified on hematoxylin and eosin stain (Figure 2C); however, interstitial spread may be subtle, and the tumor cells may be missed if CD30 immunohistochemistry is not performed. Leukemic presentation is very rare and when it is present, the tumor cells exhibit similar features to those described above and usually contain cytoplasmic vacuoles and/or few azurophilic granules (Figure 2D).
Although the morphologic variants of ALCL are not officially recognized in the ALK- group [1,36], it is still possible to make a diagnosis of ALK- ALCL in cases that have areas with a classic/common morphology adjacent to variant features, namely signet-ring cells, sarcomatoid morphology, or a background rich in neutrophils or eosinophils [14,37]. However, this may not be the case for the small cell variantand the lymphohistiocytic variant that can only be diagnosed using ALK immunohistochemistry [14,36,37]. Sarcomatoid ALCL consists of a tumor with a variable proportion of pleomorphic epithelioid cells and spindle cells arranged in short fascicles or in a storiform pattern and a myxoid background with a few scattered inflammatory cells [37,38] (Figure 3A,B). The tumor cells tend to arrange around blood vessels with partial infiltration into the vascular wall. A rare form of ALCL morphologically identical to nodular sclerosis classic Hodgkin lymphoma (CHL) is referred to as ALCL with a “Hodgkin-like pattern” [37,39] (Figure 3C,D).
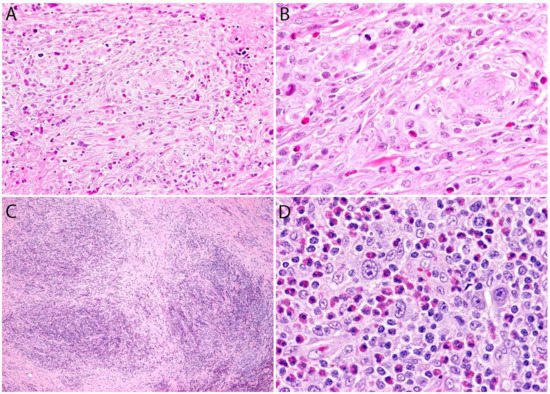
Figure 3. Variants of systemic ALK- ALCL. (A,B) Sarcomatoid ALCL. Note the perivascular arrangement and the focal areas of necrosis at the periphery. Occasional “hallmark” cells are seen. (C,D) “Hodgkin-like” ALCL. This variant is identical to nodular sclerosis classic Hodgkin lymphoma and can only be distinguished by immunohistochemistry. The tumor cells in both cases were positive for CD30, CD4, and other T-cell markers, and were negative for CD8, CD15, PAX5, and ALK (not shown in figure).
Immunohistochemistry
ALK- ALCL is strongly and diffusely positive for CD30 (>75% of cells) and negative for ALK (Figure 4A–C). CD30 decorates the lymphoma cells with a membranous and Golgi pattern (Figure 4A,B). This lymphoma shows a peculiar paradoxical expression of CD4 and of cytotoxic markers (TIA-1, granzyme B, and perforin) (Figure 4D). However, it is now well-recognized that DUSP22-rearranged cases are usually negative for cytotoxic molecules [40]. CD2, CD3, CD5, CD7, and CD8 are variably expressed (Figure 5A–C) and cases that lack all T-cell antigens are referred to as having a “null” phenotype [36,41,42]. CD56 and CD45/LCA are also variably expressed (Figure 5D), and some cases can be CD45-negative. CD43 is expressed in most cases [3,36,42]. Clusterin and MUM1/IRF4 are usually positive (Figure 5E), and in contrast to ALK+ ALCL, only ~40% of cases are positive for EMA [3] (Figure 5F). ALK- ALCL is negative for CD15, B-cell markers, and EBER ISH. However, rare cases may be positive for CD15 and/or PAX5, the latter due to extra copies of the PAX5 gene locus [43]. Unusual cytokeratin expression has also been reported in rare cases [44,45,46] (Figure 5F inset). ALK- ALCL cases with the TP63 rearrangement are positive for p63 by immunohistochemistry [40,47]. Loss of CD30 has been described in sporadic cases after the use of brentuximab-vedotin (anti-CD30) [48,49,50,51].
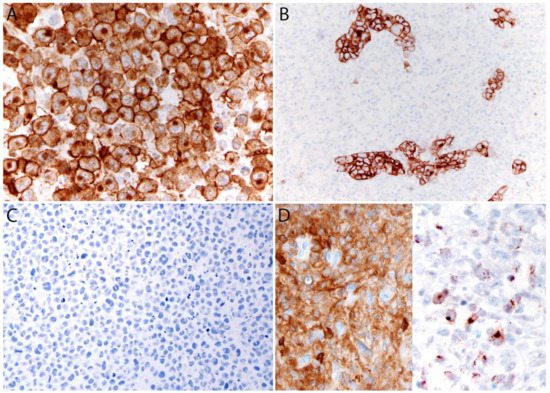
Figure 4. Immunohistochemistry in systemic ALK- ALCL. (A) CD30 (120 kDa glycoprotein) is strongly and diffusely positive in the tumor cells with a membranous and Golgi dot-like pattern. The Golgi labelling corresponds to the location of the CD30 protein precursor of 90 kDa. (B) CD30 highlights the sinusoidal distribution in a partially involved lymph node. (C) The ALK immunostain is negative. (D) Characteristic co-expression of CD4 (left) and granzyme B (right).
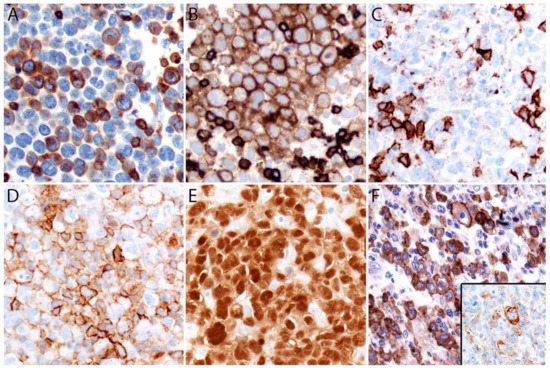
Figure 5. Immunohistochemistry in systemic ALK- ALCL. (A) Variable expression of CD3. (B) Positive CD5. (C) CD8 is negative in the lymphoma cells and positive in background cytotoxic T-cells. (D) Variable CD45. (E) Strong MUM1 expression. (F) EMA is positive in ~40% of cases. Rare cases can express ≥1 cytokeratin (F, inset) that along with a positive EMA could be misinterpreted as carcinoma if ALCL is not considered in the differential diagnosis.