Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Silvio Borrelli and Version 2 by Camila Xu.
Sodium overload is common in end-stage kidney disease (ESKD) and is associated with increased cardiovascular mortality that is traditionally considered a result of extracellular volume expansion.
- sodium
- end-stage kidney disease
- peritoneal dialysis
1. Introduction
The kidneys play a pivotal role in the body's sodium balance [1]; indeed, kidney failure is a well-known condition coupled with a remarkable sodium overload, which by inducing expansion of the extracellular volume (ECV), causes blood pressure (BP) elevation, left ventricular hypertrophy, and heart failure [2][3][2,3]. More interestingly, recent studies have highlighted that sodium accumulates in the interstitial tissue of the skin and other tissue, leading to salt-sensitive hypertension regardless of the ECV expansion [4]. Hence, restoring the sodium body balance in end-stage kidney disease (ESKD) patients represents a cornerstone of dialytic treatment but remains challenging [5].
2. Extrarenal Mechanisms for Sodium Body Balance and Hypertension
The classical two-compartment model for the body's sodium balance assumes that sodium intake is the primary determinant of ECV. According to this traditional model, in healthy humans in a steady-state, sodium intake is closely correlated to urinary sodium excretion. An abrupt increase in sodium intake, by enhancing plasma osmolarity, induces thirst and the secretion of ADH with a subsequent increase in intake and renal reabsorption of water, resulting in ECV expansion and body weight increase. In this picture, sodium excretion is finely regulated by neuro-hormonal mechanisms (the renin-angiotensin-aldosterone system and sympathetic nervous system) activated by volume and pressure sensors (Figure 1a) [1]. This model of the body's sodium balance has recently been challenged. Heer et al. showed that a high sodium intake was not associated with an increase in total body water or body weight but caused a relative fluid shift from the interstitial into the intravascular space [6]. Furthermore, a space-simulation study reported that sodium excretion changed periodically in healthy individuals with a regular intake of nutrients and, more importantly, independently of the sodium intake [7]. These findings have generated a novel hypothesis as to the internal sodium balance. According to the three-compartment model, sodium stored in the interstitial space is not osmotically inert since it activates monocyte–phagocyte system (MPS) cells by increasing local tonicity. Indeed, MPS cells sense enhanced tonicity in the interstitium by a tonicity enhancer-binding protein (ton-EBP). Activation of the ton-EBP stimulates the production and release of interleukins (e.g., monocyte chemoattractant protein-1, MCP-1, IL-6) and the vascular endothelial growth factor-C (VEGF-C), which, causing local inflammation and vascular proliferation, induces sodium clearance by the lymphatic system (Figure 1b) [8]. Several experimental studies have demonstrated that salt-sensitive hypertension may be caused by blocking the Ton-EBP/VEGF-C pathway at various levels: Ton-EBP transcription, MPS cell recruitment, VEGF receptors, and lymph vessels [9][10][11][9,10,11]. Hence, interstitial tissue could act as a sodium buffer on the MPS cells and VEGF-C-dependent lymphangiogenesis. A sodium overload may result from saturation (failure) of interstitial sodium storage.
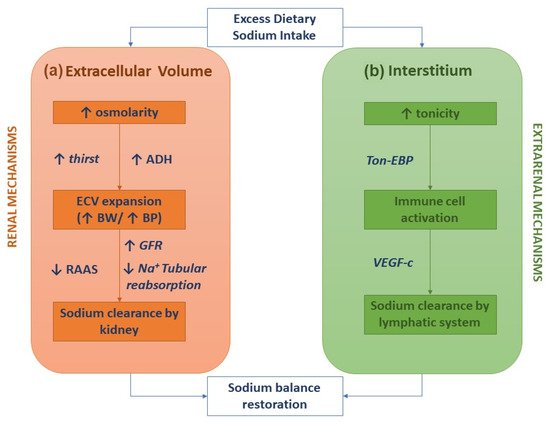
Figure 1. Sodium homeostasis according to the traditional kidney-regulated model (a) and the novel immune-regulated model (b).
Furthermore, several experimental studies have also reported an abnormal activation of the adaptive immune system (Th1 and Th17 cells) due to higher sodium exposure, thus contributing to the development of hypertension. Overall, these studies suggest a more complex link between salt-induced inflammation and hypertension [12]. According to a recent working hypothesis, a higher salt intake stimulates the dendritic cells (DC) to produce reactive oxygen species via NADPH oxidase, promoting the lipid peroxidation of fatty acids isolevuglandins. These lipid oxidation derivates to a lysine on proteins, which are in turn denatured, leading to the production of neoantigenic peptides, isolevuglandin adducts. These neoantigens activate the DCs to release proinflammatory cytokines, such as IL-6 and IL-1β. Furthermore, the DCs activated by isolevuglandin adducts stimulate the T cells to release IL-17, TNF-α, and IFN-γ, leading to hypertension and hypertension-damaged organ tissue (kidney, heart, vascular system) [13].
3. Volume-Independent Sodium Toxicity in End-Stage Kidney Disease
An optimal balance between intake and removal of sodium and water is currently considered a significant determinant of dialysis adequacy, even more than small solute clearances (urea in primis) [14][16]. Fluid overload is detected in over 50% of uremic patients treated by chronic dialysis [15][16][17,18] and contributes to their peculiar enhanced risk of cardiovascular morbidity and mortality [17][18][19,20]. However, sodium's harmful effects on the cardiovascular system in ESKD might also be independent of the ECV expansion. Indeed, the recent availability of Na23-MRI has enabled us to detect interstitial sodium storage in the skin and muscles and reveals new scenarios in the pathogenesis of sodium toxicity in ESKD. Clinical studies using Na23-MRI have shown that sodium stored in the interstitial skin tissue correlates with BP levels [19][21] and progressively increases as the GFR declines [20][21][22,23].
Interestingly, a cross-sectional study in a cohort of 99 CKD patients reported that skin sodium detected by Na23-MRI correlated with a greater left ventricular mass irrespective of the fluid overload and ambulatory BP [20][22]. This interstitial sodium concentration is even higher in the skin and the muscles of ESKD patients than in the controls and CKD patients [21][22][23][23,24,25]. Furthermore, older and African American patients on maintenance dialysis exhibit a greater sodium content than younger and non-African American ESKD patients [21][22][23,24]. Interestingly, in the study by Sahinoz et al., higher plasma inflammatory markers (IL-6 and C-reactive protein) correlated with an increased muscle and skin sodium content [22][24]. This correlation heralds an exciting link between interstitial sodium and chronic inflammation, as already reported in experimental studies [24][25][26][26,27,28]. Thus, in nephrectomized rats, exposure to two weeks of higher salt intake increased IL-6, CMP-1, and TNF-alfa levels in the dialysate. Notably, it was also associated with increased peritoneal Ton-EBP, the protein acting as a link between sodium and inflammation in the interstitial tissue. Finally, they reported that expanding the sodium intake induced peritoneal permeability (D/P creatinine increase).
Sodium Removal by Peritoneal Dialysis
Recent insights into the adverse effects of sodium stored in the skin interstitium have renewed interest in the dialytic strategies to remove sodium by dialysis [14][22][27][28][29][16,24,29,30,31]. Sodium stored in the interstitial tissue is by no means unmodifiable, given that a significant reduction in interstitial sodium has been documented after a hemodialysis session [27][29]. At the same time, higher ultrafiltration proves to be associated with lower skin sodium in patients on PD [22][24]. Furthermore, in hemodialysis patients, the use of low-sodium dialysate is associated with a lower sodium concentration in the skin and muscles [28][30]. Finally, in predialysis CKD patients, kidney transplantation caused tissue sodium accumulation to drop to the level of healthy controls [29][31].
Sodium is currently removed by convection in PD because sodium removal by diffusion is generally very slight. Thus, sodium removal must be coupled with water removal by a convective mechanism (drag solvent) to become significant. A quantitative analysis in vivo, using two liters of a glucose solution at different concentrations for 360 min dwells, estimated a sodium removal of −1.8 ± 26 mmol for 1.36%, 36.0 ± 21.0 mmol for 2.27%, and 70.5 ± 31.5 mmol for 3.86%, respectively [30][32]. Furthermore, the ultrafiltration (and sodium removal) entity depends on peritoneal permeability being significantly reduced in fast transporters [30][32].