Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ezaldeen Esawi and Version 2 by Peter Tang.
Aptamers are short single-stranded RNA or DNA oligonucleotides capable of folding into complex 3D structures, enabling them to bind to a large variety of targets ranging from small ions to an entire organism. Their high binding specificity and affinity make them comparable to antibodies, but they are superior regarding a longer shelf life, simple production and chemical modification, in addition to low toxicity and immunogenicity.
- aptamers
- drug delivery
- nanocarriers
- chemical modifications
1. Introduction
Aptamers are single-stranded RNA or DNA oligonucleotides that fold up into a distinctive 3D structure capable of binding with high affinity and specificity to small molecules up to entire organisms, with nanomolar range dissociation constants. Nucleic acid-based aptamers were first described in 1990, where the first aptamers were in vitro selected using a random library of single-stranded oligonucleotides sequences [1] by a selection procedure known as systematic evolution of ligands by exponential enrichment (SELEX). The first oligonucleotide aptamer was isolated to bind with small molecules [2]. Nowadays, the aptamer field covers various biomedical applications, including [3], therapeutic [4][5][6][4,5,6], aptasensors [7], biosensors [8][9][8,9], diagnostic [10][11][10,11], and imaging systems [12].
Aptamers need to be stabilized for in vivo use against nuclease degradation, and their small size makes them susceptible to renal filtration. Aptamers’ stabilization can be attained by chemically modifying them using different approaches. Moreover, introducing chemical modifications into nucleic acid libraries increases the interaction capabilities of aptamers and thereby their target spectrum [12]. Modified aptamers may show improved chemical diversity relative to aptamers composed entirely of natural DNA or RNA nucleotides and expand their applications in diagnostics, therapeutics, and nanotechnology [1].
Chemical modifications of aptamer oligonucleotides are needed mainly to enhance their resistance to nuclease degradation and lowering their renal filtration. Additionally, chemical modifications, in some cases, may increase the aptamer-binding affinity [13]. Many approaches have been introduced to promote the stability of aptamers without altering their binding affinity and specificity against their targets. These approaches include chemical modification of the phosphate backbone [14][15][14,15], oxygen replacement with sulfur on the ribose unit and into phosphodiester linkage, end capping at the 3′ and/or 5′ terminals [16][17][16,17], locked nucleic acids [18][19][18,19], and circular [20], multivalent, and dimerization of aptamers [21][22][21,22].
2. Aptamers and Selection Methods
Aptamer target-binding specificity and affinity are obtained using a Darwinian evolution screening technology called systematic evolution of ligands by exponential enrichment (SELEX). The SELEX methodology is quite similar for both DNA and RNA aptamers. However, RNA aptamers require an additional step of reverse transcription before amplification. Targets on which aptamer selection is conducted against are diverse, ranging from ions to whole living cells. SELEX starts by chemical synthesis of a library of random sequences of double-stranded DNA (dsDNA). The dsDNA library is either used to synthesize a single-stranded DNA (ssDNA) library or undergoes in vitro transcription to produce an ssRNA library. The resulting library sequences have the ability to fold, forming unique 3D structures. Conditions of selection, such as temperature, pH, and ionic strength, can be controlled to be compatible with the application of interest. Conventional SELEX generates aptamers by first incubating a library composed of 1013 to 1015 different folded oligonucleotide sequences with the target of choice. This library is composed of random sequences in the middle and constant regions at the 5′- and -3′ ends, which are used for primer annealing and amplification. After incubation, the bound sequences can be separated from the remaining unbound sequences, retained, and then amplified to be reintroduced in iterative selection cycles. Conditions can be changed through each cycle to achieve optimal aptamer target-binding affinity. Rounds of selection are repeated until optimal enrichment for the highest affinity and specificity aptamers are achieved. Approximately 20 rounds of selection would yield high target affinity aptamers [23][33].
Since the discovery of aptamers, a number of SELEX methods have been developed. One important variant of SELEX is the cell-based selection methodology (cell-SELEX). Cell-SELEX proved effective in developing aptamers for targets in their native status, biomarker discovery, and pathogen-infected cells. Cell-SELEX also gives an added benefit of selecting aptamers against targets existing in their original cells, which increases the specificity of selected aptamers. In cell-SELEX, the bound sequences are detached from target cells, amplified, and reintroduced to target cells in subsequent rounds of enrichment while unbound sequences are washed out. Specificity against target cells can be achieved through an extra step of counter selection, where the library is tested against control cells that are related to the target cells.
Another SELEX variant is in vivo SELEX. This method generates aptamers against targets in living organisms instead of using isolated pure targets or individual cells. This method is useful in overcoming some of the challenges facing aptamers selected by in vitro SELEX methods. The principle here is similar to that of conventional SELEX and cell SELEX. However, the library of aptamers can be injected into the peripheral vasculature of the living organism followed by tracking and isolation of specific aptamers homed into target tissues. In Vivo SELEX has been successfully applied for different diseases, including cancers and viral-infected cells [24][25][26][45,46,47].
Aptamers have many superior advantages over antibodies as the targeting ligand, including shorter production time, lower synthesis costs, better thermal stability, and a wider spectrum of targets.
3. Chemical Modifications of Aptamers and Their Impact on Pharmacological Properties
Despite the numerous encouraging characteristics of aptamers, they bear several drawbacks [27][52], such as (i) decreased biostability mainly due to rapid renal excretion and nuclease hydrolysis, (ii) nucleic acids lack functional groups that could enhance the binding affinity through extra potential interactions, and the (iii) intra-nucleotide chemical modification of aptamers dramatically affects the binding affinity.
In order to overcome these problems, modifications located at the sugar unit, the nucleobase, or the backbone of the constituting nucleotides can be introduced to enhance aptamer biostability and binding affinity [28][53]. Aptamer modification can be achieved either into the scaffold of selected aptamers through standard solid-phase synthesis (post-selection modification) or by using modified nucleoside triphosphates (NTPs) directly in the selection process [29][30][31][54,55,56]. The common chemical modification approaches of nucleic acid aptamers for the development of clinical therapeutics are discussed in the following sections.
3.1. Modifications on Nucleic Acids Terminals
3.1.1. Terminal 3′–3′ and/or 5′–5′ Internucleotide, 3′ and 5′-Biotin Conjugates
Capping the 3′-end is one of the generally used strategies to block 3′ to 5′ exonuclease attack. Capping the 3′-end with inverted deoxy-thymidine modification is usually used to increase the stability and resistance of aptamers against 3′-exonuclease in human serum [32][57]. This modification needs a modified control pore glass (CPG) with the 5′-hydroxyl of the first nucleoside attached, followed by chain elongation using the classic solid-phase phosphoramidite process [33][58].
The 3′-biotin aptamer modification is also reported to enhance stability against 3′-exonuclease (Figure 1) [32][57]. The 3′-biotin-streptavidin attached to thrombin aptamer was investigated against 3′-exonuclease activity in the blood of rodents. The results showed that 3′-biotin modification significantly enhances 3′-exonuclease resistance and decreases the clearance rate of aptamers in the blood circulation in vivo (10- to 20-fold) [16]. The 3′-biotin modified-DNA aptamer targeting severe acute respiratory syndrome (SARS) coronavirus helicase was sustained in fetal bovine serum (FBS) in double the time compared to unmodified aptamer [32][57].
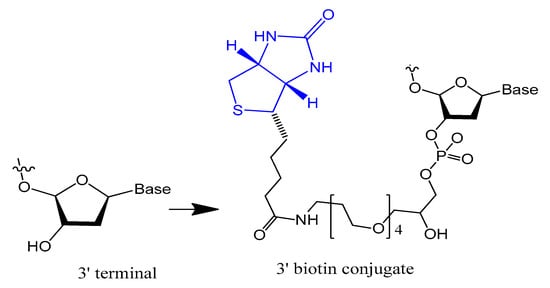
Figure 1.
Structure of the 3′-biotin conjugate.
Moreover, the stability of selected non G-quadruplex aptamer (NG8) that was modified with 3′-biotin or 3′-inverted thymidine was increased. The 3′-biotin and 3′-inverted thymidine NG8 aptamer was strongly resistant to nuclease attack in serum compared to the unmodified NG8 aptamer. The 3′-inverted thymidine aptamer remained intact for up to 72 and 31 h in 5% and 10% FBS, respectively. The 3′-inverted thymidine modification showed higher stability than the 3′-biotin modification, but both modifications were significantly more stable than the unmodified aptamer [32][57].
Riccardi et al. synthesized and characterized a dansyl-fluorescent thrombin-binding aptamer (TBA) analog, named tris-mTBA, functionalized with a 5′-biotin tag, for incorporation onto streptavidin-coated silica NPs. The results showed that tris-mTBAwas able to form an antiparallel G-quadruplex structure and retain the ability to form a duplex structure with its complementary strand (cTBA), which acts as an antidote to reverse the anticoagulation activity of TBA. Moreover, they proved that tris-mTBA inhibits the human thrombin activity 10-fold more efficiently than unmodified TBA and biotin-TBA and in a reversible manner. In addition, TBA analogs showed higher resistance to enzymatic degradation compared to the unmodified TBA due to a protective effect of the conjugating groups [34][59].
Ortigao et al. found that a minor structural change of an oligonucleotide at the two terminal internucleotide bonds, a 3′,3′ and a 5′,5′ linkage, is sufficient to stabilize these end-inverted (INV) oligonucleotides against nuclease degradation. INV oligonucleotides are degraded very slowly in biological systems, in human serum with a half-life of ~36 h, and in Xenopuslaevis oocytes with a half-life of ~10 h, whereas control "normal" oligonucleotides are completely degraded in less than 30 min in both systems [35][60].
3.1.2. 5′-End with Cholesterol and Other Lipid Moieties
Small aptamers are cleared and excreted rapidly by renal glomerular filtration. To overcome renal filtration and extend the circulation period, aptamer modifications with hydrophobic and/or bulky moiety are required [36][37][61,62]. Cholesterol was conjugated at the 5′-end of a 16-mer oligonucleotide (ODN) through a phosphate spacer, then incubated with low-density lipoprotein (LDL), leading to the formation of a cholODN-LDL. The plasma half-life of the cholODN-LDL aptamer was nearly 10 times better than the plasma half-life of the unmodified aptamer. Furthermore, the modified cholODN-LDL version showed high stability against rat serum nucleases [17].
Recently, a cholesterol-conjugated and 2′-F pyrimidine-modified RNA aptamer targeting the hepatitis C virus (HCV) NS5B protein was modified by Lee et al. This aptamer modification extended the aptamer plasma circulation time nine-fold compared to the unmodified version and enhanced the aptamer exposure to its target [38][63]. In another case, a 5′-cholesterol-modified oligonucleotide (ARC155) showed more rapid plasma clearance relative to the unconjugated aptamer, which was explained by the inability of the ARC155 folded structure to bind with plasma lipoproteins as other cholesterol-attached aptamers [36][61].
A diacylglycerol (DAG) lipid anchor was conjugated to the 5′-end of vascular endothelial growth factor (VEGF) aptamer (Figure 2). This 5′-end DAG-modified VEGF aptamer was incorporated into the bilayers of liposomes, which resulted in aptamers with improved inhibitory activity toward VEGF-induced endothelial cell proliferation in vitro and increased vascular permeability in vivo. Moreover, the residence time in plasma was considerably improved when compared to that of free aptamers [39][64].
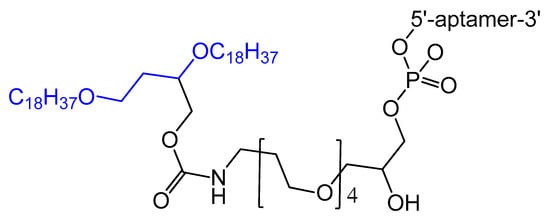
Figure 2.
Synthesis of the diacylglycerol (DAG)-modified VEGF aptamer.
A set of lipids conjugated to 5′-AS1411 aptamer (stearyl- or cholesteryl-based tails) (Figure 3) were selected and investigated for their conformational behavior and aggregation tendency in comparison with unmodified AS1411. The 5′-lipidated AS1411 derivatives folded into stable unimolecular G-quadruplex structures, forming large aggregates at a concentration of higher than 10 μM, and they maintained a similar biological behavior as unmodified aptamer with less cytotoxicity on the selected three different cancer cell lines [40][65].
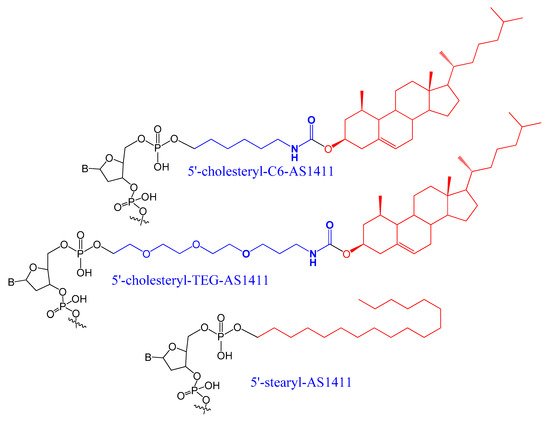
Figure 3.
A set of lipids conjugated to 5′-AS1411 aptamer (stearyl- or cholesteryl-based tails.
3.1.3. 5′-End PEGylation
The conjugation of polyethylene glycol (PEG) to drugs has been shown to increase the residence time of the drug in the body and decrease degradation by metabolic enzymes. PEG is non-toxic and nonimmunogenic and is approved by the Food and Drug Administration (FDA) [41][66].
An amino-modified spiegelmer NOX-E36 oligonucleotide was conjugated with (NHS)-ester-activated polyethylene glycol via carbodiimide coupling. This combination formula with high molecular weight PEG had the advantages of both nuclease resistance and decreased renal excretion [42][67]. MP7 is a DNA aptamer that binds to the murine extracellular domain of PD-1 (programmed death protein 1). Conjugation of MP7 DNA aptamers with large PEG molecules at the 5′ terminal via carbodiimide chemistry (Figure 4) could limit the rate of filtration and extend the half-life of this small molecule up to 24 to 48 h [37][62].
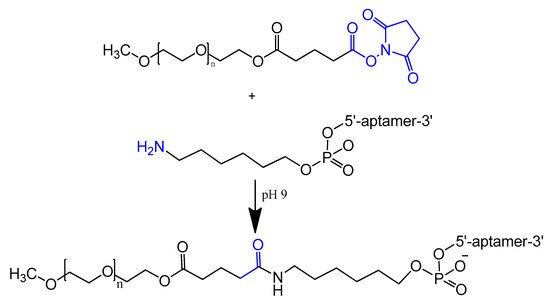
Figure 4.
Reaction scheme of aptamer conjugation to a 40-kDa polyethylene glycol (PEG) at the 5′ terminal.
An interesting new PEGylation method, sbC-PEGylation, was introduced recently for RNA aptamers acting against interleukin-17A (IL-17A) in mice and monkeys. These sbC-PEGylated aptamers were synthesized by coupling the symmetrical branching molecule 2-cyanoethyl-N,N-diisopropyl phosphoramidite to the 5′ end of the aptamer, before conjugating two PEG molecules to the aptamer. The sbC-PEGylated aptamers had improved pharmacokinetic properties and showed excellent stability in the blood circulation. Moreover, one of the sbC-PEGylated aptamers, 17M-382, inhibited interleukin-6 (IL-6) in a concentration-dependent manner, with the IC50 of 17M-382 two times lower than that of non-PEGylated 17M-382 [43][68].
The bifunctionalized anti-MUC1 aptamer (NH(2)-AptA-SR) was conjugated with different PEG types (either a conventional branched PEG or the comb-shaped polyPEG) to enhance its biodistribution properties against MCF-7 cell lines. The body clearance data showed that more than 60% of the un-PEGylated aptamer was excreted after 5 h compared to 43% to 51% in the case of the newly modified aptamers [44][69].
The PEGylation of doxorubicin-attached anti-MUC1 aptamer (PEG-APT-DOX) increases the targeting efficacy of the aptamer by reducing the non-specific uptake of doxorubicin by RAW 264.7 macrophages. PEG-APT-DOX kept more than 80% of the RAW cells viable while killing more than 60% of the MCF7 cells. This proves the desirable cytotoxic effect of doxorubicin to MCF7 cells was not hindered by the modification [45][70].
3.2. Modifications on the Sugar Ring
3.2.1. 2′-Substitutions
Modifications on the 2′ position of the sugars were effective at improving the aptamer serum half-life. The 2′-Fluoro (2′-F), 2′-amino (2′-NH2) and 2′-O-methyl (2′-OMe) are the most common 2′-substitute modifications on the ribose unit. Usually, it is used to increase nuclease resistance and optimize aptamer affinity (Figure 5) [46][47][48][23,24,71].
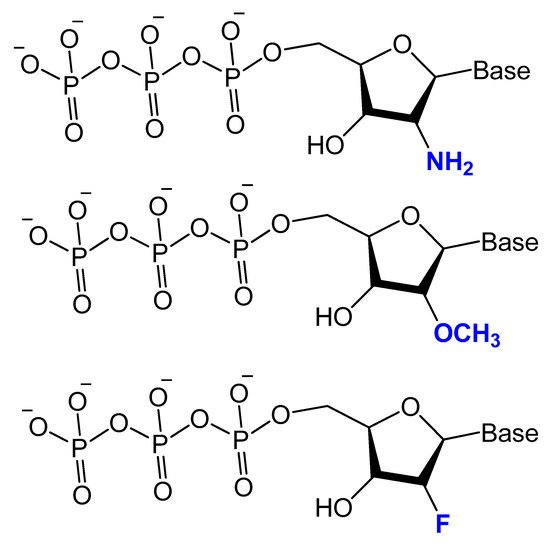
Figure 5. Chemical structures of 2′-modified nucleotides used in selection experiments to generate aptamers with enhanced pharmacokinetic properties.
For example, two 2′-F-modified thrombin-binding aptamers (PG13 and PG14) showed approximately a four-fold increased binding affinity to thrombin and up to seven-fold higher nuclease resistance. The G-quadruplex stability of the modified aptamer was increased up to 48-fold in 10% FBS [13]. Lin et al. developed a 2′-NH2 group-modified RNA aptamer against human neutrophil elastase (HNE). This modified aptamer showed a good binding affinity and 10-fold enhanced stability in human serum and urine compared to unmodified aptamer [49][72]. High-affinity 2′-amino-2′-deoxypyrimidine-modified RNA ligands have been reported to be potent inhibitors of basic fibroblast growth factor (bFGF). Compared to unmodified RNA with the same sequence, 2′-aminopyrimidine ligands are at least 1000-fold more stable in 90% human serum [30][55]. 2′-Amino-modified RNA and DNA aptamers that bind to vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) have been investigated by Green et al. They showed nuclease resistant properties with high binding affinity [50][73].
Despite 2′-methoxy modification being one of the post-selection procedures, due to bulky 2′-OMe-NTPs [51][81], a combination of three 2′-OMe-NTPs was used in a SELEX experiment to generate an aptamer that binds to the polypeptide tissue factor pathway inhibitor (TFPI). This modified aptamer showed high selectivity and binding affinity to correct thrombin generation hemophilia A and B [52][82].
These 2′-modified aptamers can be easily conjugated, as unmodified aptamers, to different nanocarriers loaded with certain chemotherapeutic agents. For example, 2′-OMe-modified aptamers conjugated to a polymeric nanoparticles loaded with docetaxel, an anticancer agent, showed a high specificity as well as a targeted toxicity improvement [53][83].
A 2′-deoxy-2′-fluoro-modified arabinonucleotide (2′F-ANA) was investigated based on a thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG), an anti-HIV phosphorothioate PS-d(TTGGGGTT), and a DNA telomeric sequence d(GGGGTTTTGGGG) by UV thermal denaturation (Tm) and circular dichroism (CD) experiments. The results showed that the replacement of deoxyguanosines that adopt the anti-conformation (antiguanines) with 2′F-arabino guanosines can stabilize G-quartets and maintain the quadruplex conformation while replacement of syn-guanines with 2′F-arabino guanosines is not favored and results in a dramatic switch to an alternative quadruplex conformation. In addition, the data showed that the appropriate incorporation of 2′F-ANA residues into G-quadruplexes leads to an increase in the melting temperature of the complex formed. Moreover, the nuclease resistance of 2′F-ANA-modified thrombin-binding aptamers was increased up to 48-fold in 10% FBS, with a 4- to 5-fold enhancement in binding affinity to thrombin [13]. Similarly, Wilds and Damha used UV thermal melting and CD experiments to discover the thermodynamic stability and helical conformation of 2′F-ANA/RNA and 2′F-ANA/DNA hybrids. They showed that 2′F-ANA had enhanced RNA affinity to RNase H enzyme relative to that of DNA and phosphorothioate DNA. The 2′F-ANA modification also showed favorable pairing to single-stranded DNA [54][84].
The stability of 2′-deoxy-2′-fluoroarabinonucleic acid (2′F-ANA) to hydrolysis has been investigated under acidic and basic conditions. 2′F-ANA was found to have increased stability compared to both DNA and RNA in enzyme-free simulated gastric fluid (pH ~1.2). Under basic conditions, 2′F-ANA also showed good stability. Furthermore, phosphorothioate-2′F-ANA linkage was found to be much more susceptible to enzymatic cleavage than the phosphorothioate-DNA [55][85].
The differential stability of 20 F-ANA RNA and ANA RNA hybrid duplexes was evaluated by NMR and theoretical calculations. An increased binding affinity of 2′ F-ANA was observed due to a favorable pseudo hydrogen bond (2′ F–purine H8), which contrasts with unfavorable 2′-OH–nucleobase steric interactions in the case of ANA. The 2′ F-ANA strand′s structure was more compatible with the A-like structure of a hybrid duplex and more suitably reorganized for duplex formation [56][86].
3.2.2. 4′-Oxygen Replacement
Replacing the 4′-oxygen atom of the sugar unit with a sulfur atom is rarely utilized in selection experiments of aptamer isolation (Figure 6) [57][87]. Synthesized 4′-thiouridine (4′-thio-UTP) and 4′-thiocytidine (4′-thio-CTP) triphosphates were used in the in vitro selection of anti-thrombin thioRNA aptamers. The 4′-thio-modified aptamer showed a high affinity (KD = 4.7 nM), with a 50-fold increase in resistance to RNase A [58][88]. Minakawa et al. isolated fully modified 4′-thioRNA aptamers against human alpha-thrombin using four types of 4′-thioribonucleoside triphosphates (4′-thioNTPs). The modified aptamer displayed a similar binding affinity to thrombin as the partially modified aptamer (KD = 7.2 nM) [57][87].
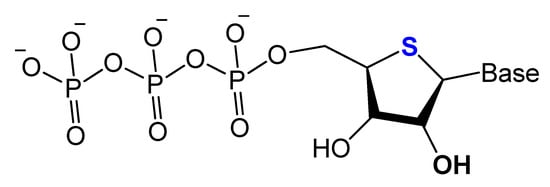
Figure 6.
Structure of 4′-thioNTPs.
3.2.3. Locked and Unlocked Nucleic Acid
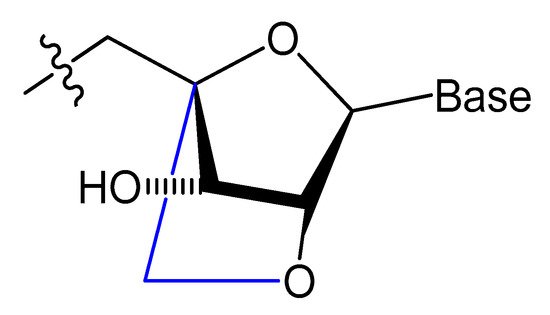
A methylene linkage between 2′-O and 4′-C of the sugar ring produces an analog of ribonucleotide called locked nucleic acid (LNA) (Figure 7). This modification showed a better thermostability and vastly enhanced nuclease resistance [18][19][18,19].
LNA/DNA chimera LNA5, forming a stable complex against HIV-1 trans-activating response (TAR) RNA, was synthesized from a shortened and stable version of the hairpin RNA aptamer identified by in vitro selection against TAR. The results indicated that these modifications provide good protection towards nuclease digestion in bovine serum and keep the same binding affinity of the unmodified RNA aptamer [59][89]. Shi et al. developed another LNA/DNA chimeric aptamer probe through proper LNA incorporation and 3′-3′-thymidine (3′-3′-T) capping (Figure 8). The serum stability of the modified TD05 aptamer, a DNA aptamer against lymphoma Ramos cells, was gradually enhanced by 10-fold, with maintained affinity and specificity to Ramos cells [60][90].
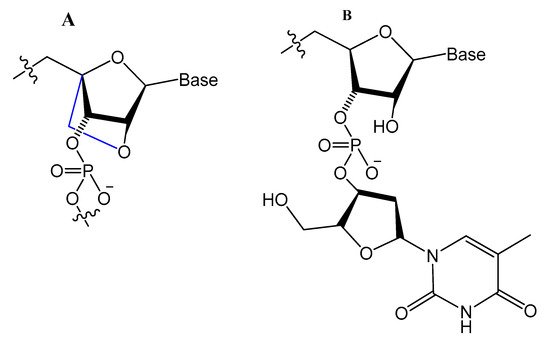
A Tenascin-C-binding aptamer was modified with LNA nucleotides (TTA1) that exhibited improved plasma stability and maintained strong binding to Tenascin-C [61][91]. Moreover, an avidin-binding DNA aptamer was modified systematically with LNA and a 2′-amino derivative as demonstrated in Figure 9. At certain positions, the modified aptamer actually showed improved binding affinity (KD value of 4.20 nM) [62][92].
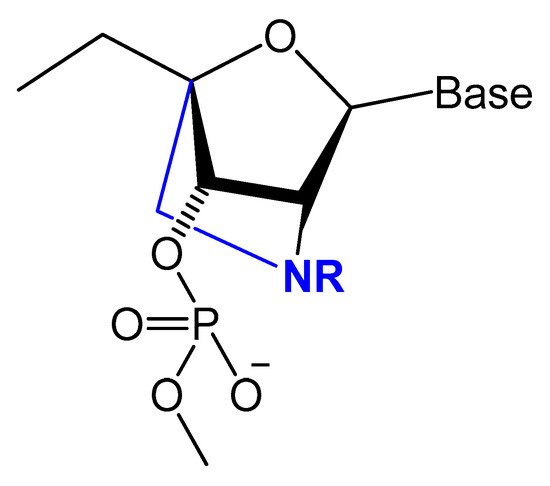
Figure 9.
Structures of LNA and 2′-amino-LNA nucleotide monomers.
Unlocked nucleic acid (UNA) is another modification in the ribose unit, achieved by eliminating the single bond between C2′ and C3′ of the sugar. Such a modification makes the aptamer more flexible. This structure flexibility may ease the strain in tight aptamer loops (Figure 10) [12]. The 15-mer thrombin-targeted DNA that underwent UNA modifications on the loop regions showed increased thermodynamic stability, with significant aptamer affinity and anticoagulant efficiency [63][93].
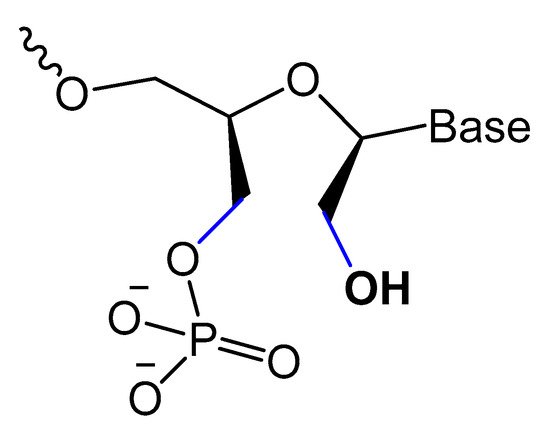
A thrombin-binding quadruplex aptamer with a UNA-modified thrombin-binding aptamer (UNA-modified TBA) was developed. It was found that UNA substitution in the loops of the quadruplex could increase the binding affinity and clotting time in blood samples. The UNA monomer is allowed in many positions of the aptamer without significantly changing the thrombin-binding properties [63][93].
Aptamers could be selected with LNA or UNA in their structure, which may give better results. Three different LNA-nucleoside triphosphates, LNA-TTP, LNA-ATP, and LNA-5-methyl-CTP, were tested as substrates for KOD DNA polymerase. The results showed that KOD DNA polymerase is good for the synthesis of DNA oligonucleotide duplexes containing LNA nucleotides [64][94].
The effect of the presence of nucleosides in unlocked nucleic acid (UNA), locked nucleic acid (LNA), or β-l-RNA series, as analogs to RE31-DNA aptamer for effective prolonging of the thrombin time, was evaluated by Kotkowiak et al. They showed that all modified residues can influence the thermal and biological stabilities of G-quadruplex in a position-dependent manner. The aptamers modified simultaneously with UNA and LNAs possess a two-fold higher anticoagulant effect. RE31 variants modified with nucleosides in UNA, LNA, or β-l-RNA series exhibited prolongation of aptamer stability in human serum [65][95].
3.3. Modifications on the Phosphodiester Linkage
3.3.1. Methylphosphonate or Phosphorothioate
One of the common aptamer modifications can be achieved by replacing the phosphodiester linkage with methylphosphonate or phosphorothioate on the α-phosphorous (Figure 11) [14][27][14,52].
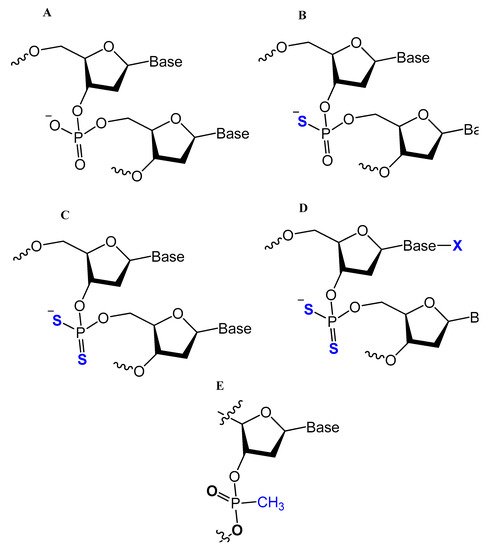
The phosphorothioate modification might influence the thermal stability of the quadruplex structure in different G-quadruplex-forming oligonucleotides, and the phosphorothioate backbone is considered to be responsible for strongly binding and inhibiting the gp120 envelope protein of the HIV [14]. Partial thiophosphorylated substitutions with maximum thermal stability were selected for evaluating their stabilities under conditions of nuclease RQ1 DNAse hydrolysis and their antithrombin activities in blood plasma. A promising modified oligonucleotide (GGTTGGTGTGGTTGG), with the structure modified only in TT loops (LL11), retained thrombin-binding aptamer properties with high resistance to biodegradation [67][96]. A thrombin-binding aptamer, d(GGSTSTSGGTGTGGSTSTSGG), with thio-substitutions in both TT loops exhibited a similar antithrombin efficiency compared to the unmodified one, with better resistance to DNA nuclease in blood serum [15]. Abeydeera et al. reported that phosphorodithioate (PS2) substitution on a single nucleotide of RNA aptamers improved the target binding affinity by 1000-fold by stabilizing the phosphate backbone [68][97].
Post-SELEX modifications by the addition of short phosphorothioate caps to the 3′- and 5′-ends of the 2′-amino-modified RNA and DNA aptamers against VPF/VEGF showed high binding affinity and increased nuclease resistance [50][73].
Different types of phosphorothioate aptamers have been isolated via in vitro combinatorial selection. For example, King et al. selected purified recombinant human NF-kappa B proteins, RelA(p65) and p50, duplex thioaptamers. These phosphorothioate aptamers showed high affinity besides competitive binding with the duplex 22-mer-binding site, Ig kappa B [69][99].
Somasunderam et al. utilized the same protocol to isolate phosphorothioate aptamers acting as inhibitors of the RNase H domain of HIV-1 reverse transcriptase with high affinity (KD of 70 nM) [70][100]. They also separated monothiophosphate-modified aptamers that specifically bind to CD44 with a high affinity in the range 180–295 nM, an affinity significantly higher than that of hyaluronic acid [71][101]. The selection of a thioaptamer targeting the Dengue virus type-2 envelope (DENV-2) protein domain III was also achieved by Gandham et al. DENTA-1 thioapatamer binds to DENV-2 EDIII, with a dissociation constant of 154 nM [72][102].
Recently, thiophosphate ester aptamers (TA), selected from large combinatorial libraries, with CD44 (CD44TA) targeting moiety, was attached to discoidal silicon mesoporous microparticles (SMP) to improve the accumulation of these carriers in infected macrophages in the lungs. This thioaptamer significantly lowered the bacterial load in the lungs, caused recruitment of T lymphocytes, and enhanced binding affinity and specificity for proteins as well as stability in vitro [73][74][103,104].
X-aptamers were the next generation of phosphorothioate aptamers, in which a C5 position of the nucleobase was modified with a drug-like functionality [75][105]. This modification showed a significant enhancement of nuclease resistance and increased binding affinities [76][106]. The best X-aptamer modification with small molecule drugs and antibodies can be achieved if these molecules can fold into unique 3-D structure scaffolds to bind specifically to the target protein [66][50].
Prater and Miller reported a single methylphosphonateinternucleotide linkage at the 3′-end of an oligo-2′-O-methylribonucleotide (Figure 12) that showed high binding affinities for their complementary targets and prevented degradation by the 3′-exonuclease activity found in mammalian serum [77][107].
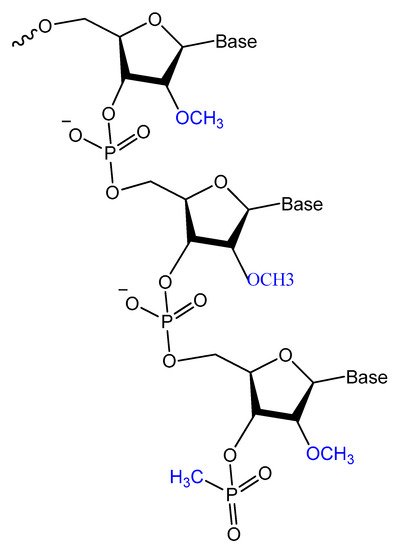
Figure 12.
General structure of oligo-2′-
O
-methylribonucleotide containing a single methylphosphonate linkage at the 3′ end.
A comparison of duplex stabilities between different phosphorothioate, methylphosphonate, and 2′-OCH3 RNA analogs of two self-complementary DNA 14-mers was conducted by Kibler-Herzog et al. Highly modified phosphorothioates or methylphosphonates are less stable than their partially modified counterparts, which are less stable than the unmodified parent compounds. Phosphorothioate derivatives were found to be more stable when the linkage modified was between adenines rather than between thymines [78][108].
The effect of chemical modifications on the thermal stability of different G-quadruplex-forming oligonucleotides was investigated by Sacca et al. The methylphosphonate-modified 15mer oligonucleotide, known as the thrombin-binding aptamer (15TBA-M) gave no observable thermal transition, resulting in a flat thermal profile. In contrast, the unmodified oligonucleotide, phosphorothioate aptamers (15TBA-S), and the 2′-O-methyl ribonucleotide analogs (15TBA-O) gave a reversible and concentration-dependent thermal transition. In addition, loss of the negative charge at the level of the phosphate backbone, as in the methylphosphonate analogs, leads to a strong destabilization of the G-quadruplex structure [14].
3.3.2. Triazole Modification
Oligonucleotide triazole modification instead of phosphodiester linkage has been investigated extensively in many studies [79][80][109,110] to protect the oligonucleotides from nuclease hydrolysis [81][111].
This modification is usually achieved using a click reaction between azide- and alkyne-bearing nucleosides (Figure 13) [82][83][112,113] or through automated phosphoramidite synthesis with modified dinucleoside blocks (Figure 14) [84][114].
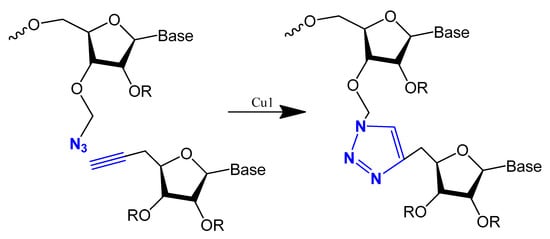
Figure 13. The copper-catalysed alkyne–azide cycloaddition reaction between an azide and a terminal alkyne to produce a 1,4-triazole.
Figure 14.
Triazol-modified thymidine dinucleotides.
The triazole unit can link nucleotides directly or with methylene or single ether bond linkage. These analogs are similar to oligonucleotides and show increased resistance to nuclease cleavage. The triazole oligonucleotide analogs demonstrated DNA binding affinities similar to those of unmodified oligonucleotides. The modification was shown to protect oligonucleotides from nuclease hydrolysis [81][111].
Triazole-modified DNA aptamers with a structure similar to thrombin-inhibiting G-quadruplexes, TBA15 and TBA31, had been tested for their stabilities and binding affinities. No change was observed in their binding affinities, but the triazole modification protected aptamers from nuclease hydrolysis and increased their stabilities [80][85][110,115].
3.4. Modifications on the Bases and SOMAmers
The most modified positions on nucleic acid bases occur on pyrimidines’ C5-position and the N7 position of purines (Figure 15). These sites have been shown to be in good contact with polymerase enzymes and are easily adapted in the major groove of nucleic acid duplexes [86][116].
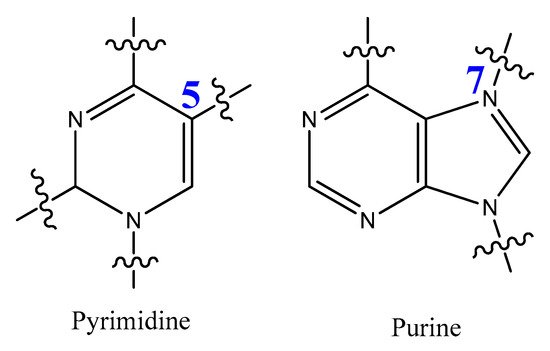
Figure 15.
Positions of pyrimidine and purine modification.
These modifications on nucleotides involve, for example, the coupling of l-proline-containing residues, dipeptide, urea derivative, and a sulfamide residue, followed by triphosphorylation. These modified 2′-deoxyribonucleoside triphosphates, dNTPs, were shown to be excellent substrates to be incorporated into DNA by the polymerase chain reaction (PCR) and are excellent candidates for SELEX [87][117].
Modified base aptamers are able to retain target binding properties, and thus they may enhance the binding affinity [88][89][118,119]. For example, a base-modified aptamer, 5-(1-pentynyl)-2′-deoxyuridine, used instead of thymidine, was isolated via a selection experiment against human coagulation protease thrombin (Figure 16) [88][118].
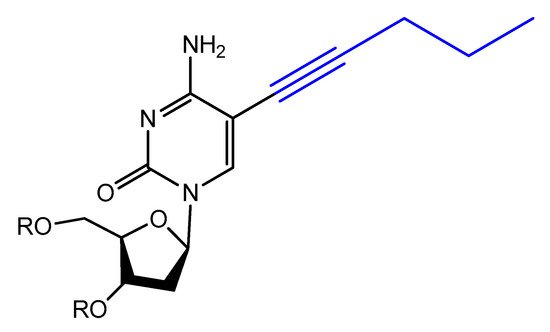
Figure 16.
Structure of modified 5-(1-pentynyl)-2′-deoxyuridine used in aptamer selection.
Gupta et al. introduced a different chemical modification by adding new side chains at the 5-position of uracil. These side chains ranged from high hydrophilic to more hydrophobic fragments. They assessed the impact of these side chains on the plasma pharmacokinetics of the modified aptamers.
These changes were effective in increasing the chemical diversity of the aptamers. By increasing the rate of discovery of high-affinity ligand to protein targets, they also caused an increase in nuclease resistance, with lower renal clearance for more hydrophilic side chains [89][119].
Photo-reactive chromophore 5-iodo-UTP was incorporated in a SELEX to generate a base-modified aptamer with a high capability for covalent interaction with HIV-1 Rev protein [90][120]. An anti-fibrinogen base aptamer modified with boronic thymidine-5′-triphosphate (Figure 17) was isolated by Li et al. This aptamer has specific recognition of fibrinogen glycosylation, enhancing the binding affinity compared to unmodified aptamer [91][121].
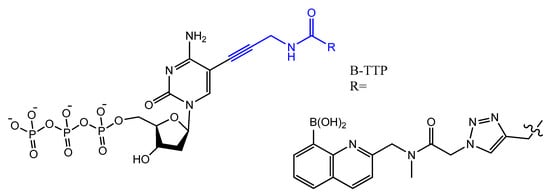
Figure 17.
The chemical structures of B-TTP.
The addition of an adenine residue to the C5 position of uracil ((E)-5-(2-(N-(2-(N6-adeninyl)ethyl)) carbamylvinyl)-uracil) increased the hydrogen bonding interaction and enhanced its efficiency to target the anticancer agent, camptothecin derivative 1 (CPT1). A very potent aptamer, CMA-70, was selected, and then improved to the shorter (CMA-59) aptamer. An improved binding affinity was seen for both modified aptamers compared to the natural aptamers [92][122]. Moreover, enantioselective base-modified aptamers isolated by SELEX were capable of binding only to the (R)-isomer of thalidomide. The aptamer thymidine was replaced with a modified deoxyuridine with a cationic group via a C5 hydrophobic methylene linker. The additional functional group improved the stability against nucleases and increased the binding affinity to thalidomide [93][123]. An arginine-modified dUTP (Figure 18) was involved in a SELEX experiment to improve its enantioselectivity. The isolated aptamers displayed enantioselective binding to the negatively charged glutamic acid as the target [94][124].
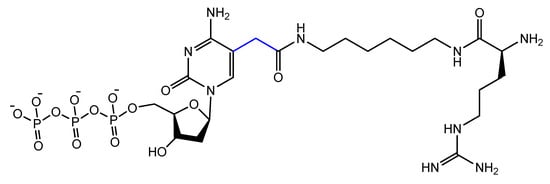
Figure 18.
Chemical structure of the arginine-modified analog of dUTP.
A glycol-DNA aptamer was produced from an alkyne unit, 5-ethynyl-modified dUTP, via SELMA selection could recognize the monoclonal antibody, 2G12, which is known to bind to mannose-rich glycans on the HIV envelope protein, gp120, thus neutralizing various HIV strains [95][96][97][125,126,127].
Lee and colleagues revealed that 5-BzdU (5-(N-benzylcarboxyamide)-2-deoxyuridine) modification of the AS1411 aptamer might selectively increase its targeting affinity to cancer cells while having no significant influence on the normal healthy cells [98][128]. The 5-BzdU residue was further modified by replacing the benzyl group by other aromatic or aliphatic groups to enhance the binding affinity of this modified aptamer to their targets [99][129].
Another increasingly expanding approach utilizes the replacement of natural nucleotides with artificial unnatural bases in the DNA sequence to improve the therapeutic properties [100][101][130,131]. A nucleoside triphosphate modified with a tyrosine-like phenol (Figure 19) was used in the selection of DNA aptamers against Escherichia coli DH5α cells. The modified aptamer displayed high selectivity and affinity for the target cells compared to the unmodified aptamer [102][132].
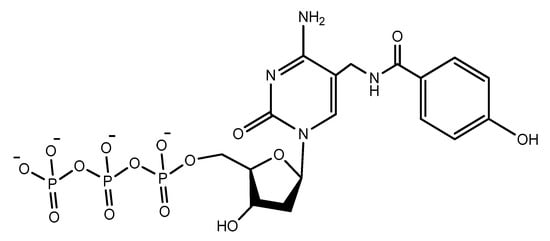
Figure 19.
The modified phenol-dUTP nucleotide.
5-[(p-Carborane-2-yl)ethynyl]-2′-deoxyuridine 5′-O-triphosphate was synthesized and used by Balintová et al. as substrate for KOD XL DNA polymerase in a primer extension (PEX) reaction to generate carborane-modified DNA or oligonucleotides. These carborane-modified hydrophobic aptamers may increase the potential interactions against hydrophobic proteins or analytes [103][133]. C5-modified carboxamide pyrimidines’ functionality was a smart choice to facilitate the attachment of other hydrophobic groups, such as benzene, thiophene, naphthalene, isopropyl, and amino acid derivatives (Figure 20) [104][134].
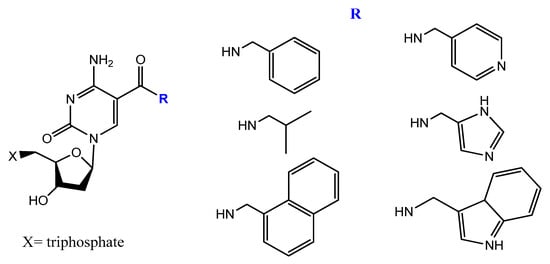
A new protocol was lately described to select nucleobase-modified aptamers. This protocol utilizes click chemistry (CuAAC) to introduce the favored nucleobase modification based on alkyne-modified uridine (5-ethynyl-deoxyuridine (EdU)) instead of thymidine. This new protocol enables a wide range of functionality and generates modified DNA aptamers with extended interaction properties [105][135].
The slow off-rate-modified aptamers (SOMAmers) are aptamers with significant base modification to give a protein-like functionality. This formulation improves the binding affinities and binding kinetics with enhanced selectivity when compared to traditional aptamers. This is achieved by increasing both the number and strength of the hydrophobic interactions between nucleic acids and the corresponding targets, thus partially mimicking the binding mode of antibodies and other proteins. The power of this kind of base modification is that it exhibits very little nuclease degradation over a 48-h incubation in human serum [106][107][136,137], it facilitates the detection of various proteins in the blood serum, and it has been widely applied in the discovery of disease biomarkers [108][109][138,139].
Modified DNA SOMAmers ((5-(N-benzylcarboxamide)-2′-deoxyuridine (Bn-dU) or 5-[N-(1-naphthylmethyl)carboxamide]-2′-deoxyuridine (NapdU) replacing dT) that inhibit interleukin-6 (IL-6) signaling, a key component of inflammatory diseases, were found to be stable in serum and blocked the interaction of IL-6 with its receptor, IL-6Rα [106][136].
An advanced SOMAmer-based assay was developed for quantification of soluble glypican-3 in hepatocellular carcinoma (HCC) patient samples using glypican-3 SOMAmer. The assay verified its good sensitivity, accuracy, and precision compared to the traditional antibody-based assay, with a high binding affinity [110][140]. Gawande and co-workers explored selection experiments using double-modified DNA aptamers with amino-acid-like moieties on pyrimidine bases to target proprotein convertase subtilisin/kexin type 9. They isolated aptamers that showed higher affinity, biostability, and inhibitory potency compared to singly modified aptamers with broad utility in research, diagnostic, and therapeutic applications [111][141].
Wang et al. reported a biophysical and enzymatic properties study of three widely used protein-like side chain dNTPs: 8-histaminyl-deoxyadenosine (dAimTP), 5-guanidinoallyl-deoxyuridine (dUgaTP), and 5-aminoallyl-deoxycytidine (dCaaTP). The base-pairing abilities of oligonucleotides having one or three modified nucleosides were tested by thermal denaturation analysis and as a substrate for enzymatic polymerization with both modified and natural dNTPs [112][142].
3.5. Spiegelmers
Spiegelmers are the synthetic mirror image of d-nucleic acids that show high resistance to nuclease degradation and may retain their binding affinity to their d-form targets or be selected with high binding affinity to new targets (Figure 21) [113][143]. For example, NOX-A12, a structured mirror image RNA oligonucleotide in the l-configuration that neutralizes stromal cell-derived factor-1, interferes with chronic lymphocytic leukemia migration and drug resistance [114][144]. NOX-A12, a spiegelmer that binds and neutralizes CXCL12, was developed for interference with CXCL12 in the tumor microenvironment and for cell mobilization.
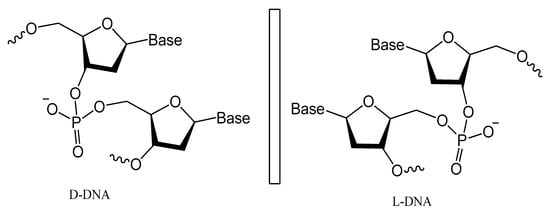
Figure 21.
Structures of
l
-deoxyoligonucleotide (
l
-DNA). Mirror image aptamers are composed of non-natural
l
-ribose nucleotides.
An l-RNA aptamer targeting the HIV-1 trans-activation responsive (TAR) RNA was developed. This spiegelmer showed great specificity and strong binding activity based on tertiary interactions more than Watson–Crick pairing [115][145]. In addition, NOX-G15 is a mixed DNA/RNA mirror image aptamer that binds to the glucagon and improves glucose tolerance in models of type 1 and type 2 diabetes [116][146].
A 67-mer l-enantiomeric spiegelmer for gonadotropin-releasing hormone (GnRH) was selected from a random pool of oligonucleotides, and this effective antagonist spiegelmer showed a high binding affinity (KD = 20 nM) with longer plasma half-life stability [117][147]. Another l-GnRH spiegelmer was chemically synthesized according to the isolated natural d-GnRH aptamer. The resulting spiegelmer had similar affinities to that of d-aptamers [118][148]. A biologically stable mirror image enantiomeric l-DNA spiegelmer against bacterial Staphylococcal enterotoxin B was developed. The spiegelmer bound the whole protein target, with only a slightly reduced affinity, which shows the possibility of identifying spiegelmers against large protein targets [119][149]. Spiegelmers also undergo similar different strategies and chemical modifications as natural aptamers to enhance their stability against nucleases and improve their binding affinity [113][143]. A nuclease-resistant modified l-RNA aptamer (MLRA) with cationic nucleotide, 5′ aminoallyl-uridine, was isolated in an in vitro selection process and this spiegelmer was capable of binding oncogenic pre-miR-19a with exceptional affinity, and the cationic modification was absolutely crucial for binding [120][150].
Finally, Taylor and Holliger described protocols for the replication of artificial analogs of DNA and RNA having a different backbone or sugar homologous xeno nucleic acids (XNAs). For the directed evolution of synthetic oligonucleotide ligands (XNA aptamers) for specific targeting of proteins or nucleic acid units, a cross-chemistry selective exponential enrichment (X-SELEX) approach is used. This approach may be applied to select and isolate fully modified XNA aptamers for a wide range of target molecules [121][151].
Conventional SELEX, based on only four natural DNA/RNA nucleotides, often yields poor binders only. Synthetic biology has increased the number of DNA/RNA building blocks, with tools to sequence, PCR amplifies, and clone artificially expanded genetic information systems (AEGISs). Several examples have been reported of a SELEX using AEGIS, producing a molecule that binds to cancer cells