Hyperuricemia is related with cardiovascular risks. Reactive oxygen species (ROS) are produced simultaneously with the formation of uric acid by xanthine oxidases. Intracellular uric acid has also been reported to promote the production of ROS. The ROS and the intracellular uric acid itself regulate several intracellular signaling pathways, and alterations in these pathways may result in the development of atherosclerotic lesions.
- Hyperuricemia
- Atherosclerosis
1. Introdution
Hyperuricemia is a common metabolic syndrome. Elevated uric acid levels are risk factors for gout, hypertension, and chronic kidney diseases. Furthermore, various epidemiological studies have also demonstrated an association between cardiovascular risks and hyperuricemia. In hyperuricemia, reactive oxygen species (ROS) are produced simultaneously with the formation of uric acid by xanthine oxidases. Intracellular uric acid has also been reported to promote the production of ROS. The ROS and the intracellular uric acid itself regulate several intracellular signaling pathways, and alterations in these pathways may result in the development of atherosclerotic lesions.
2. The Role of Hyperuricemia in the Pathogenesis of Atherosclerosis
2.1. Oxidative Stress
-
ROS are produced due to the increased activity of xanthine oxidase in the metabolic process of uric acid;
-
The expression and activity of NADPH oxidase increase;
-
Mitochondrial ROS (mtROS) are produced due to mitochondrial injury.
2.1.1. Xanthine Oxidoreductase
2.1.2. NADPH Oxidase
2.1.3. Mitochondrial ROS
2.2. Inflammatory Signaling Pathway
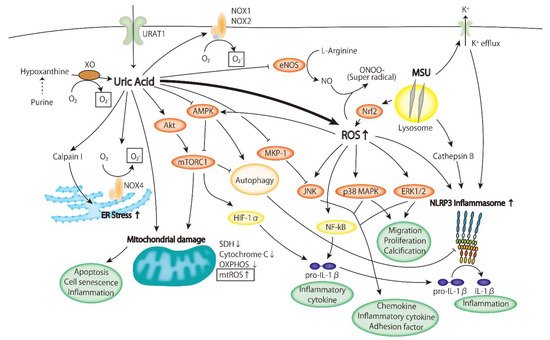
2.2.1. ERK/p38 MAPK Cascade
2.2.2. AMPK
2.2.3. PI3K-Akt Pathway
2.2.4. Inflammasome
3. The Impact Cof Uric Acidnclusion
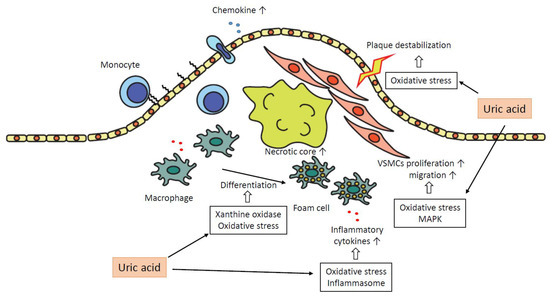
In summary, uric acid plays a pro-atherogenic role in several steps in the progression of plaques as follows. Uric acid promotes oxidative stress and destabilization of NO, which leads to vasoconstriction and endothelial dysfunction. The expressions of chemokines, such as MCP-1, are increased, and monocytes are recruited into the subendothelial layer. Macrophages in subendothelial are differentiated into foam cells depending on oxidative stress by uric acid and the effect of xanthine oxidase. These foam cells or macrophages secrete inflammatory cytokines, and uric acid promotes the production of the cytokines. The inflammatory cytokines attract further inflammatory cells and bring the formation of the necrotic core. Uric acid promotes proliferation and migration of VSMCs via activation of MAPK and oxidative stress, which leads to the progression of atheromatous plaque. Oxidative stress derived from mitochondrial dysfunction by uric acid results in the destabilization of plaques. Inflammation augmented by uric acid via activation of inflammasomes or several inflammatory signaling pathways contributes to the development of atherosclerosis in each atherogenic step (Figure 2).
References
- Sevanian, A.; Davies, K.J.; Hochstein, P. Serum Urate as an Antioxidant for Ascorbic Acid. Am. J. Clin. Nutr. 1991, 54 (Suppl. S6), 1129S–1134S.
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of Xanthine Oxidoreductase and NAD(P)H Oxidase in Endothelial Superoxide Production in Response to Oscillatory Shear Stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297.
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of Uric Acid and Xanthine Oxidase in Atherosclerotic Plaque. Am. J. Cardiol. 2001, 88, 188–191.
- Doehner, W.; Schoene, N.; Rauchhaus, M.; Leyva-Leon, F.; Pavitt, D.V.; Reaveley, D.A.; Schuler, G.; Coats, A.J.S.; Anker, S.D.; Hambrecht, R. Effects of Xanthine Oxidase Inhibition with Allopurinol on Endothelial Function and Peripheral Blood Flow in Hyperuricemic Patients with Chronic Heart Failure: Results from 2 Placebo-Controlled Studies. Circulation 2002, 105, 2619–2624.
- Butler, R.; Morris, A.D.; Belch, J.J.; Hill, A.; Struthers, A.D. Allopurinol Normalizes Endothelial Dysfunction in Type 2 Diabetics with Mild Hypertension. Hypertension 2000, 35, 746–751.
- Guthikonda, S.; Sinkey, C.; Barenz, T.; Haynes, W.G. Xanthine Oxidase Inhibition Reverses Endothelial Dysfunction in Heavy Smokers. Circulation 2003, 107, 416–421.
- El Solh, A.A.; Saliba, R.; Bosinski, T.; Grant BJ, B.; Berbary, E.; Miller, N. Allopurinol Improves Endothelial Function in Sleep Apnoea: A Randomised Controlled Study. Eur. Respir. J. 2006, 27, 997–1002.
- Linas, S.L.; Whittenburg, D.; Repine, J.E. Role of Xanthine Oxidase in Ischemia/Reperfusion Injury. Am. J. Physiol. 1990, 258 Pt 2, F711–F716.
- Granger, D.N. Role of Xanthine Oxidase and Granulocytes in Ischemia-Reperfusion Injury. Am. J. Physiol.-Heart Circ. Physiol. 1988, 255, H1269–H1275.
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine Oxidoreductase in Atherosclerosis Pathogenesis: Not Only Oxidative Stress. Atherosclerosis 2014, 237, 562–567.
- Kushiyama, A.; Okubo, H.; Sakoda, H.; Kikuchi, T.; Fujishiro, M.; Sato, H.; Kushiyama, S.; Iwashita, M.; Nishimura, F.; Fukushima, T.; et al. Xanthine Oxidoreductase Is Involved in Macrophage Foam Cell Formation and Atherosclerosis Development. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 291–298.
- Cheung, K.J.; Tzameli, I.; Pissios, P.; Rovira, I.; Gavrilova, O.; Ohtsubo, T.; Chen, Z.; Finkel, T.; Flier, J.S.; Friedman, J.M. Xanthine Oxidoreductase Is a Regulator of Adipogenesis and PPARgamma Activity. Cell Metab. 2007, 5, 115–128.
- Martinez-Hervas, S.; Real, J.T.; Ivorra, C.; Priego, A.; Chaves, F.J.; Pallardo, F.V.; Viña, J.R.; Redon, J.; Carmena, R.; Ascaso, J.F. Increased Plasma Xanthine Oxidase Activity Is Related to Nuclear Factor Kappa Beta Activation and Inflammatory Markers in Familial Combined Hyperlipidemia. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 734–739.
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine Oxidoreductase Regulates Macrophage IL1β Secretion upon NLRP3 Inflammasome Activation. Nat. Commun. 2015, 6, 6555.
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. Biomedicines NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206.
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse Effects of the Classic Antioxidant Uric Acid in Adipocytes: NADPH Oxidase-Mediated Oxidative/Nitrosative Stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596.
- Chao, H.H.; Liu, J.C.; Lin, J.W.; Chen, C.H.; Wu, C.H.; Cheng, T.H. Uric Acid Stimulates Endothelin-1 Gene Expression Associated with NADPH Oxidase in Human Aortic Smooth Muscle Cells. Acta Pharmacol. Sin. 2008, 29, 1301–1312.
- Choi, Y.-J.; Shin, H.-S.; Choi, H.S.; Park, J.-W.; Jo, I.; Oh, E.-S.; Lee, K.-Y.; Lee, B.-H.; Johnson, R.J.; Kang, D.-H. Uric Acid Induces Fat Accumulation via Generation of Endoplasmic Reticulum Stress and SREBP-1c Activation in Hepatocytes. Lab. Investig. 2014, 94, 1114–1125.
- Yang, Y.; Zhou, Y.; Cheng, S.; Sun, J.L.; Yao, H.; Ma, L. Effect of Uric Acid on Mitochondrial Function and Oxidative Stress in Hepatocytes. Genet. Mol. Res. 2016, 15.
- Sánchez-Lozada, L.G.; Lanaspa, M.A.; Cristóbal-García, M.; García-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.A.; Kang, D.-H.; Johnson, R.J. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron. Exp. Nephrol. 2012, 121, e71–e78.
- Kimura, Y.; Yanagida, T.; Onda, A.; Tsukui, D.; Hosoyamada, M.; Kono, H. Soluble Uric Acid Promotes Atherosclerosis via AMPK (AMP-Activated Protein Kinase)-Mediated Inflammation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 570–582.
- Su, Y.; Hu, L.; Wang, Y.; Ying, G.; Ma, C.; Wei, J. The Rho Kinase Signaling Pathway Participates in Tubular Mitochondrial Oxidative Injury and Apoptosis in Uric Acid Nephropathy. J. Int. Med. Res. 2021, 49.
- Muslin, A.J. MAPK Signalling in Cardiovascular Health and Disease: Molecular Mechanisms and Therapeutic Targets. Clin. Sci. 2008, 115, 203–218.
- Kırça, M.; Oğuz, N.; Çetin, A.; Uzuner, F.; Yeşilkaya, A. Uric Acid Stimulates Proliferative Pathways in Vascular Smooth Muscle Cells through the Activation of P38 MAPK, P44/42 MAPK and PDGFRβ. J. Recept. Signal Transduct. 2016, 37, 167–173.
- Li, Z.; Shen, Y.; Chen, Y.; Zhang, G.; Cheng, J.; Wang, W. High Uric Acid Inhibits Cardiomyocyte Viability Through the ERK/P38 Pathway via Oxidative Stress. Cell. Physiol. Biochem. 2018, 45, 1156–1164.
- Kanellis, J.; Watanabe, S.; Li, J.H.; Kang, D.H.; Li, P.; Nakagawa, T.; Wamsley, A.; Sheikh-Hamad, D.; Lan, H.Y.; Feng, L.; et al. Uric Acid Stimulates Monocyte Chemoattractant Protein-1 Production in Vascular Smooth Muscle Cells Via Mitogen-Activated Protein Kinase and Cyclooxygenase-2. Hypertension 2003, 41, 1287–1293.
- Xin, Y.; Wang, K.; Jia, Z.; Xu, T.; Xu, Q.; Zhang, C.; Liu, J.; Chen, R.; Du, Z.; Sun, J. Zurampic Protects Pancreatic β-Cells from High Uric Acid Induced-Damage by Inhibiting URAT1 and Inactivating the ROS/AMPK/ERK Pathways. Cell. Physiol. Biochem. 2018, 47, 1074–1083.
- Nomura, J.; Busso, N.; Ives, A.; Tsujimoto, S.; Tamura, M.; So, A.; Yamanaka, Y. Febuxostat, an Inhibitor of Xanthine Oxidase, Suppresses Lipopolysaccharide-Induced MCP-1 Production via MAPK Phosphatase-1-Mediated Inactivation of JNK. PLoS ONE 2013, 8, e75527.
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. AMP-Activated Protein Kinase Promotes Macrophage Polarization to an Anti-Inflammatory Functional Phenotype. J. Immunol. 2008, 181, 8633.
- Cordero, M.D.; Williams, M.R.; Ryffel, B. AMP-Activated Protein Kinase Regulation of the NLRP3 Inflammasome during Aging Implication of AMPK in Aging. Trends Endocrinol. Metab. 2018, 29, 8–17.
- Ma, A.; Wang, J.; Yang, L.; An, Y.; Zhu, H. AMPK Activation Enhances the Anti-Atherogenic Effects of High Density Lipoproteins in ApoE -/- Mice. J. Lipid Res. 2017, 58, 1536–1547.
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin Inhibits Monocyte-to-Macrophage Differentiation via AMPK-Mediated Inhibition of STAT3 Activation: Potential Role in Atherosclerosis. Diabetes 2015, 64, 2028–2041.
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andrés-Hernando, A.; Ishimoto, T.; Sánchez-Lozada, L.G.; et al. Counteracting Roles of AMP Deaminase and AMP Kinase in the Development of Fatty Liver. PLoS ONE 2012, 7, e48801.
- Cicerchi, C.; Li, N.; Kratzer, J.; Garcia, G.; Roncal-Jimenez, C.A.; Tanabe, K.; Hunter, B.; Rivard, C.J.; Sautin, Y.Y.; Gaucher, E.A.; et al. Uric Acid-Dependent Inhibition of AMP Kinase Induces Hepatic Glucose Production in Diabetes and Starvation: Evolutionary Implications of the Uricase Loss in Hominids. FASEB J. 2014, 28, 3339.
- Zhang, Y.; Yamamoto, T.; Hisatome, I.; Li, Y.; Cheng, W.; Sun, N.; Cai, B.; Huang, T.; Zhu, Y.; Li, Z.; et al. Uric acid induces oxidative stress and growth inhibition by activating adenosine monophosphate-activated protein kinase and extracellular signal-regulated kinase signal pathways in pancreatic β cells. Mol Cell Endocrinol. 2013, 375, 89.
- Luo, C.; Lian, X.; Hong, L.; Zou, J.; Li, Z.; Zhu, Y.; Huang, T.; Zhang, Y.; Hu, Y.; Yuan, H.; et al. High Uric Acid Activates the ROS-AMPK Pathway, Impairs CD68 Expression and Inhibits OxLDL-Induced Foam-Cell Formation in a Human Monocytic Cell Line, THP-1. Cell. Physiol. Biochem. 2016, 40, 538–548.
- García-Arroyo, F.E.; Monroy-Sánchez, F.; Muñoz-Jiménez, I.; Gonzaga, G.; Andrés-Hernando, A.; Zazueta, C.; Juárez-Rojas, J.G.; Lanaspa, M.A.; Johnson, R.J.; Sánchez-Lozada, L.G. Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats. Biomolecules 2019, 9, 601.
- Zhao, Y.; Qian, Y.; Sun, Z.; Shen, X.; Cai, Y.; Li, L.; Wang, Z. Role of PI3K in the Progression and Regression of Atherosclerosis. Front. Pharmacol. 2021, 12, 263.
- Fernández-Hernando, C.; Ackah, E.; Yu, J.; Suárez, Y.; Murata, T.; Iwakiri, Y.; Prendergast, J.; Miao, R.Q.; Birnbaum, M.J.; Sessa, W.C. Loss of Akt1 Leads to Severe Atherosclerosis and Occlusive Coronary Artery Disease. Cell Metab. 2007, 6, 446.
- Crişan, T.O.; Cleophas, M.C.P.; Novakovic, B.; Erler, K.; van de Veerdonk, F.L.; Stunnenberg, H.G.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Uric Acid Priming in Human Monocytes Is Driven by the AKT-PRAS40 Autophagy Pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 5485–5490.
- Hu, Y.; Zhao, H.; Lu, J.; Xie, D.; Wang, Q.; Huang, T.; Xin, H.; Hisatome, I.; Yamamoto, T.; Wang, W.; et al. High Uric Acid Promotes Dysfunction in Pancreatic β Cells by Blocking IRS2/AKT Signalling. Mol. Cell. Endocrinol. 2021, 520, 111070.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 97.
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847.
- Klauser, A.S.; Halpern, E.J.; Strobl, S.; Gruber, J.; Feuchtner, G.; Bellmann-Weiler, R.; Weiss, G.; Stofferin, H.; Jaschke, W. Dual-Energy Computed Tomography Detection of Cardiovascular Monosodium Urate Deposits in Patients with Gout. JAMA Cardiol. 2019, 4, 1019–1028.
- Abdellatif, W.; Chow, B.; Nicolaou, S. THU0598 ROLE OF DUAL-ENERGY CT AS A SCREENING TOOL FOR CORONARY GOUT. Ann. Rheum. Dis. 2019, 78 (Suppl. S2), 590–592.
- Yokose, C.; Eide, S.; Simeone, F.; Shojania, K.; Nicolaou, S.; Becce, F.; Choi, H.K. Frequently Encountered Artifacts in Novel Application of Dual-Energy CT to Vascular Imaging: A Pilot Study—ACR Meeting Abstracts. Arthritis Rheumatol. 2019, 71 (Suppl. S10). Available online: https://acrabstracts.org/abstract/frequently-encountered-artifacts-in-novel-application-of-dual-energy-ct-to-vascular-imaging-a-pilot-study/ (accessed on 11 November 2021).
- Andrés, M.; Quintanilla, M.-A.; Sivera, F.; Sánchez-Payá, J.; Pascual, E.; Vela, P.; Ruiz-Nodar, J.-M. Silent Monosodium Urate Crystal Deposits Are Associated with Severe Coronary Calcification in Asymptomatic Hyperuricemia: An Exploratory Study. Arthritis Rheumatol. 2016, 68, 1531–1539.
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884.
- Cavalcanti, N.G.; Marques, C.D.L.; Lins, T.; Pereira, M.C.; Rêgo, M.J.B.D.M.; Duarte, A.L.B.P.; Pitta, I.D.R.; Pitta, M.G.D.R. Cytokine Profile in Gout: Inflammation Driven by IL-6 and IL-18? Immunol. Investig. 2016, 45, 383–395.
- Ruggiero, C.; Cherubini, A.; Ble, A.; Bos, A.J.; Maggio, M.; Dixit, V.D.; Lauretani, F.; Bandinelli, S.; Senin, U.; Ferrucci, L. Uric Acid and Inflammatory Markers. Eur. Heart J. 2006, 27, 1174–1181.