Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Frederic Relaix and Version 2 by Yvaine Wei.
Skeletal muscle development and regeneration rely on the successive activation of specific transcription factors that engage cellular fate, promote commitment, and drive differentiation. Emerging evidence demonstrates that epigenetic regulation of gene expression is crucial for the maintenance of the cell differentiation status upon division and, therefore, to preserve a specific cellular identity. This depends in part on the regulation of chromatin structure and its level of condensation. Chromatin architecture undergoes remodeling through changes in nucleosome composition, such as alterations in histone post-translational modifications or exchange in the type of histone variants.
- myogenesis
- histone variants
- PAX7
- MYOD
- H3.3
- HIRA
- H2A.Z
- macroH2A
1. Introduction
1.1. Establishment of the Skeletal Muscle Lineage
Skeletal muscle tissue has a high regenerative potential that relies on tissue-specific stem cells, the satellite cells. These cells are characterized by the expression of the paired-homeobox transcription factor PAX7 and, in a subset of muscles, by the co-expression of its paralog PAX3 [1]. Muscle stem cells are established during fetal development where they adopt a satellite position under the basal lamina of the fiber and progressively initiate cell cycle exit into quiescence [2]. A pool of PAX3- and PAX7-double positive muscle stem cells constitutes a reservoir that allows fetal and postnatal muscle growth [3][4][5][3,4,5]. In the trunk and limb muscles, PAX3/7 lie genetically upstream of the myogenic regulatory factors (MRFs) that trigger cell commitment towards the myogenic lineage. The early-expressed MRFs include MYF5, MRF4 (MYF6), and MYOD, while Myogenin (MYOG) is expressed later during myoblast differentiation and is associated with terminal cell cycle exit [2]. The signaling cascades and the gene regulatory networks that operate to establish the myogenic lineage have been studied thoroughly but the epigenetic regulation of such processes is less understood.
1.2. The Epigenetic Regulation of Gene Expression
Skeletal muscle development and regeneration depend on the fine regulation of transcription factor activity and subsequent gene expression. The epigenetic status of a specific gene locus constitutes additional layers of regulatory events that control cell differentiation, fate, and identity. The epigenetic regulation of gene expression depends on the chromatin architecture, the histone post-translational modifications (PTMs), the nucleosome composition, and DNA methylation. In eukaryotic cells, nucleosomes are composed of 147 bp of DNA that wraps around the histone octamer complex, which contains a central tetramer comprising the histones H3-H4 and two external dimers of histones H2A-H2B [6][7][6,7]. The nucleosome is further stabilized by the presence of the linker histone H1. N-terminal tails of the histones and the C-terminal tail of the histone H2A extend beyond the nucleosome complex and are subjected to PTMs [8]. Histone tails can undergo PTMs such as acetylation, methylation, phosphorylation, and ubiquitylation, among others. Nucleosome remodeling is linked to several cellular processes such as DNA replication, DNA damage repair, transcriptional regulation, and maintenance of constitutive heterochromatin (centromere and telomeres) [6]. Based on these nuclear phenomena, the incorporation of histones into nucleosomes is characterized as being cell cycle dependent or independent, with distinct histone variants being deposited at each process. In mice, H2A, H2B, and H3 present several variants, while H4 has only one [6].2. The Role of the Non-Canonical h3 Histone Variant H3.3 in Myogenesis
2.1. The Histone H3 Family
The histone H3 family comprises two canonical, replication-dependent histone variants, H3.1 and H3.2, the centromeric-specific histone H3 variant CENPA, and the non-canonical histone variant H3.3. The replication-independent histone H3.3 differs from H3.1 and H3.2 by 5 and 4 amino acids (aa), respectively [7][9][7,19]. H3.3 incorporation into chromatin was identified as a requirement for the epigenetic memory of an active gene by stabilizing gene expression in somatic cells upon division [10][20]. In gene bodies H3.3 correlates with the H3K36me3 histone mark and is broadly associated with active transcription. Moreover, H3.3 enrichment in promoters and regulatory regions was mainly associated with active transcription in different cell types, given its preferential association with active histone marks like H3K4me3 and H3K9ac [11][21]. However, recent studies found that the recruitment of H3.3 into chromatin is associated with transcription regulation, either to activate or repress gene expression, when enriched in enhancers or promoters [12][22]. In mouse embryonic stem cells (mESC) the H3.3 histone variant is specifically deposited into the nucleosomes by the histone chaperone HIRA (gene bodies and transcription start sites (TSS)) and by DAXX in constitutive heterochromatin (telomeres and pericentromeric regions) [6][9][6,19]. Interestingly, recruitment of H3.3 to transcription factor binding sites (TFBS) is mostly independent of HIRA, which suggests involvement of other chaperones in the regulation of transcription [13][14][23,24].
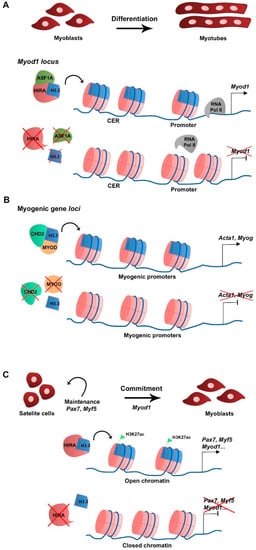
2.2. H3.3 Function during Myogenesis
In skeletal muscle cells H3.3 plays a major role in cell differentiation. In primary cultures of chick myoblasts, the synthesis of the canonical histone variant H3.2 is predominant to that of H3.3 in proliferating culture conditions, consistent with H3.2 being replication-dependent. When triggered to differentiate, myoblasts stop replicating, fuse into myotubes, and H3.2 synthesis becomes undetectable while H3.3 continues to be synthesized, suggesting that H3.3 is required during myogenic differentiation [15][25]. At the RNA level, canonical H3.1 and H3.2 variant-encoding genes are expressed in myoblasts in growth culture conditions but not in differentiation, while H3.3-encoding genes are expressed in both growth and differentiating conditions [16][26]. In the mouse myoblast cell line C2C12, H3.3 incorporation in the promoter and the core enhancer region (CER) of Myod1 is required for Myod1 expression in differentiating myoblasts [16][26]. In addition, the histone chaperone HIRA and its cofactor ASF1A mediate H3.3 recruitment to the Myod1 locus. The loss of H3.3, HIRA, or ASF1A leads to decreased Myod1 expression levels and blocks the differentiation potential of the myoblasts that fail to fully differentiate and form myotubes (Figure 1A). Mechanistically, the enrichment of H3.3 at Myod1 regulatory regions promotes the transition towards a more permissive chromatin state that facilitates the recruitment of the RNA polymerase II [16][26]. The HIRA-ASF1A complex further regulates myogenic differentiation by interacting with the muscle-specific transcription factor MEF2, which is required for the activation of MEF2 target gene expression [17][27]. However, putative changes in histone variant incorporation into chromatin were not addressed in this context. The phosphorylation of HIRA by the AKT1 kinase was proposed to mediate HIRA function as an H3.3 chaperone [18][28]. The levels of phosphorylated HIRA are high in proliferating C2C12 myoblasts, which limits the expression of myogenic genes, while dephosphorylation is required for H3.3 deposition and gene expression activation upon differentiation [18][28]. Interestingly, MYOD regulates myogenic gene expression by interacting with the ubiquitous chromodomain helicase DNA-binding domain 2 (CHD2) protein to deposit H3.3 in myogenic gene loci prior to differentiation in C2C12 [19][29]. The knockdown of Chd2 or Myod1 prevents H3.3 incorporation in myogenic genes but not in housekeeping genes (Figure 1B) [19][29]. This suggests that tissue-specific transcription factors play a role in the epigenetic regulation of target gene transcription by recruiting ubiquitous chromatin regulators. CHD2 is also part of the H3.3 incorporation complex in developmentally regulated gene loci of mESC, being essential to prevent suppressive chromatin formation at these genomic regions. In Chd2-depleted mESC, there is a decreased H3.3 deposition and increased enrichment of the histone repressive mark H3K27me3 at developmental gene loci [20][30]. Consequently, there is a blockage in mESC differentiation into the different germ layers. These studies indicate that CHD2 and H3.3 are associated with tissue differentiation in both pluripotent and somatic cells.Figure 1. Schematic representation of H3.3 function in myogenesis. (A) At the myoblast stage Myod1 expression is regulated by H3.3 deposition in the vicinity of the CER and promoter by the HIRA-ASF1A complex (Yang et al., 2011). (B) To allow differentiation and myotube formation, CHD2 directly interacts with MYOD to deposit H3.3 in promoters of myogenic differentiation-related genes and activate transcription (Harada et al., 2012). (C) Upon muscle injury, activated satellite cells require H3.3 deposition by HIRA in myogenic gene regulatory regions to maintain cell identity (Pax7, Myf5 expression) and commitment (Myod1). H3.3 enrichment correlates with that of H3K27ac and with an open chromatin state, which favors myogenic gene expression (Esteves de Lima et al., 2021).
3. H2A Histone Variants and Myogenic Gene Expression
3.1. The H2A Family
The histone variants from the H2A family display higher variation among their sequences compared to the H3 family. The H2A family is composed of several replication-independent variants besides the canonical H2A, such as H2A.Z, H2A.X, H2A.Bbd, and macroH2A (mH2A) [7][21][7,49].
H2A.Z histone variant is encoded by two genes, H2afz and H2afv, that originate two distinct protein isoforms differing by 3 aa, H2A.Z-1, and H2A.Z-2, respectively [22][50]. The H2A.Z histone variant is mostly enriched at TSS, promoters, enhancers, facultative heterochromatin, and centromeres, and was linked to both transcriptional activation and repression, in which H2A.Z-associated PTMs play a major role [23][51]. H2A.Z is expressed in C2C12 myoblasts and silencing its expression does not interfere with myoblast differentiation or myotube formation, which shows that its continued expression is not required for differentiation [24][52]. A non-acetylatable form of H2A.Z (where the 5 tail lysines are mutated into arginines) fused to GFP (H2A.Z-Ac-mut-GFP) can be incorporated into the genome in a similar fashion as the wild type H2A.Z fused to GFP (H2A.Z-GFP), including at the Myod1 and Myog loci (Figure 2A). H2A.Z-Ac-mut-GFP-expressing myoblasts display reduced myogenic gene expression, such as Myod1, Myog, and Myh3, when triggered to differentiate. Impaired myogenesis is associated with the lack of RNA polymerase II recruitment in the presence of H2A.Z-Ac-mut-GFP to myogenic gene loci upon differentiation [24][52]. Consequently, H2A.Z-Ac-mut-GFP overexpression in C2C12 cells blocks myotube formation while H2A.Z-GFP overexpression does not significantly interfere with this process, which shows the H2A acetylation regulates differentiation [24][52]. This shows that acetylation of the histone variant H2A.Z plays a role in transcription initiation of myogenic gene expression. The exchange of H2A for its variant H2A.Z is modulated by the chromatin remodeling complex SNF2-related CBP activator protein (SRCAP), a mechanism conserved in yeast and in mammals [25][26][27][53,54,55]. ChIP-seq analysis of C2C12 cells confirmed the presence of p18Hamlet (ZNHI1), a component of the SRCAP complex and a substrate of the p38 MAPK pathway, at the Myog promoter in differentiating C2C12 cells [25][53]. In addition, p18Hamlet is required for the incorporation of H2A.Z at the Myog promoter, and the enrichment of these two proteins at this genomic region increases during differentiation of C2C12 cells and murine primary myoblasts. Phosphorylation of p18Hamlet by p38 is required for its recruitment, and for the incorporation of H2A.Z in the Myog promoter, which suggests that p38 MAPK-dependent signals can impact on chromatin structural changes [25][53]. Inhibition of the expression of components of the SRCAP complex leads to impaired myogenic gene expression and blocks myoblast differentiation [25][53]. Myofibroblast differentiation that relies on TGF-β1 expression is also regulated by H2A.Z occupancy [28][56]. In contrast, in this case, in order to facilitate TGF-β1 expression, H2A.Z must be depleted from the TGF-β1 promoter region through a mechanism that requires the ribosomal function regulator ELF6 (Yang et al., 2015).
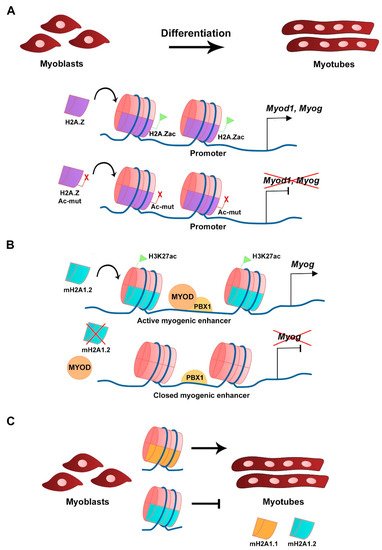
Figure 2. Histone H2A variants in myogenesis. (A) Acetylation of H2A.Z variant at the CER is required for Myod1 expression and myoblast differentiation. Overexpression of a mutated and non-acetylatable form of H2A.Z inhibits Myod1 expression (Law and Cheung, 2015). (B) The mH2A1.2 variant is required for myogenic enhancer activation prior to differentiation and correlates with H3K27ac histone mark. mH2A1.2 enrichment allows MYOD-PBX1 complex formation at the Myog promoter, activating transcription (Dell’Orso et al., 2016). (C) Distinct mH2A1 isoforms have different roles on myoblast differentiation (Hurtado-Bagès et al., 2020).
3.2. The mH2A Family
The mH2A subfamily of histone variants is about three times the size of the canonical H2A due to the presence of an evolutionary conserved non-histone globular macrodomain, described to be associated with X chromosome inactivation, transcriptional repression, and reprogramming inhibition [29][30][31][32][60,61,62,63]. The transcriptional repression activity is associated with increased stability of heterotypic mH2A-H2B-containing nucleosomes compared with canonical H2A-H2B-containing nucleosomes [33][64]. Three distinct mH2A histones have been identified, mH2A1.1 and mH2A1.2, which are two isoforms generated from alternative splicing of the H2afy gene, and mH2A2 that is encoded by H2afy2 [34][35][36][65,66,67]. RNA-seq analysis revealed that in C2C12 myoblasts the mH2A1.2 isoform is more expressed than mH2A1.1 in growth conditions, while in myotubes both isoforms are expressed at similar levels [37][68]. The comparable levels of expression for both isoforms in myotubes is the consequence of a switch in splicing that occurs after myoblast differentiation and leads to the decrease of mH2A1.2 transcript levels and the increase in mH2A1.1 [38][69]. Silencing of mH2A1.2 with siRNAs does not affect C2C12 myoblasts in growth conditions but inhibits Myog expression and myotube formation when triggered to differentiate [37][68]. Moreover, gene ontology (GO) analysis of RNA-seq data revealed terms associated with muscle cell development and differentiation to be downregulated in mH2A1.2 siRNA-transfected C2C12 cells. In the same silencing conditions, ChIP-seq analysis showed that there is a specific loss in the enrichment of the active transcription-associated histone mark H3K27ac in myogenic-specific promoters and enhancers, which shows the requirement of mH2A1.2 at these loci to maintain H3K27 acetylation and active gene expression (Figure 2B) [37][68]. The recruitment of the homeodomain-containing transcription factor PBX1, required for the MYOD-dependent activation of Myog expression, to muscle development-related gene loci is also regulated by mH2A1.2 prior to differentiation (Figure 2B) [37][68]. The mH2A1 and mH2A2 double knockout mice have impaired prenatal and postnatal development, which is associated with the mH2A function in regulating metabolic-related gene expression in the liver [39][70]. Moreover, mice lacking the histone variant mH2A1 are viable and fertile [40][71]. In both cases, analyses to address skeletal muscle defects in these mice have not been described. However, when myoblasts obtained from muscles of mH2A1-null mice are cultured in vitro, they differentiate and express MYH3 but lack the ability to form large myotubes [41][72]. Both mH2A1 isoforms regulate myoblast fusion, however with distinct outcomes (Figure 2C) [41][72]. Specific inhibition of each of the mH2A1 isoforms in C2C12 cells with siRNAs revealed that while mH2A1.1 promotes myoblast fusion, mH2A1.2 inhibits it [41][72]. This phenotype is linked to the opposite regulation of genes associated with GO terms such as extracellular matrix organization, cell adhesion, and skeletal system development. ChIP-seq analysis identified the mH2A1.1 isoform to be enriched at fusion-related genes when C2C12 are triggered to differentiate, which links mH2A1.1 to transcription activation and myoblast fusion [41][72].