Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Camila Xu and Version 2 by Camila Xu.
Tocotrienols (T3s), members of the vitamin E family, are natural compounds found in various food sources and exist as four naturally occurring analogues known as alpha (α), beta (β), delta (δ), and gamma (γ).
- tocotrienols
- colorectal cancer
- cell lines
- biomarkers
- Cytoscape
- PRISMA
- KEGG
- STRING
1. Introduction
Colorectal cancer (CRC) is the world’s third most prevalent malignancy and the fourth leading cause of cancer mortality, with almost 1.4 million new cases and approximately 700,000 deaths reported annually [1]. Considering its prevalence, by 2030, the CRC burden is projected to rise by 60%, to over 2.2 million new cases and 1.1 million deaths [2]. More than two thirds of all patients and about 60% of CRC-related deaths are found in countries with a high or extremely high human development index (HDI) [1,2][1][2].
Tocotrienols (T3s), members of the vitamin E family, are natural compounds found in various food sources and exist as four naturally occurring analogues known as alpha (α), beta (β), delta (δ), and gamma (γ). The general chemical structure of tocotrienols is shown in Figure 1. Each analogue has different functional groups that are denoted by R1, R2, and R3 [3]. In α-tocotrienol, R1, R2, and R3 represent methyl (Me) groups, but in β-tocotrienol, R1, R2, and R3 represent Me, hydrogen (H), and Me groups, respectively. In γ-tocotrienol, R1 represents H while R2 and R3 represent Me groups. However, in δ-tocotrienol, R1 and R2 represent the H group while R3 stands for the Me group [3]. The T3s are unsaturated and possess an isoprenoid side-chain, which allows them to efficiently penetrate tissues with saturated fatty layers. The anticancer potential of T3s is only beginning to receive recognition as several mechanistic studies have shown that T3s have unique anticancer properties, which are modulated through several cancer-associated mechanisms and pathways (Figure 1) [3[3][4],4], such as inhibition of telomerase activity through suppression of protein kinase C (PKC) activity in cancer cells [5]; and blocked expression of hypoxia-induced vascular endothelial growth factor (VEGF), interleukin-8 (IL-8), and cyclooxygenase 2 (COX-2), which play critical roles in cancers as these act as autocrine growth factors for carcinogenesis and tumor neovascularization [6,7,8][6][7][8]. On the contrary, T3s are reported to enhance the expression of p21 and p27, both known to cause cell cycle arrest [9]. In addition, T3s can induce apoptosis in cancer cells through several mechanisms, which includes inhibition of the nuclear factor-κB (NF-κB) pathway and its regulated gene products [10] as well as regulation of both intrinsic and extrinsic apoptotic pathways by modulating the caspase cascades, expression of B cell lymphoma 2 (BCL2), BCL2-associated X protein (BAX), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by upregulating the expression of death receptors (DRs) [11,12][11][12].
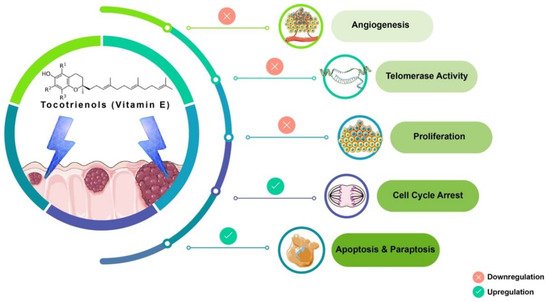
Figure 1. Anticancer actions of tocotrienols. Tocotrienols have been reported to exert anticancer effects by several mechanisms, such as induction of cell death via apoptosis and paraptosis in cancer cells, cell cycle arrest, inhibition of proliferation, suppression of the expression of the human telomerase reverse transcriptase (hTERT) in cancer cells as well as inhibition of angiogenesis. [α-tocotrienol: R1 = Me, R2 = Me, R3 = Me; β-tocotrienol: R1 = Me, R2 = H, R3 = Me; γ-tocotrienol: R1 = H, R2 = Me, R3 = Me; δ-tocotrienol: R1 = H, R2 = H, R3 = Me.
Tocotrienol is a form of vitamin E that is gaining recognition for its immense benefits against various types of cancers and other diseases. The anticancer effects of T3s shown in Figure 1 only represent a small portion of the overall T3-based research. The current state of information on this lesser-known type of vitamin E calls for more investigations to garner more scientific evidence that could further strengthen T3s’ many functions and attributes in various experimental models.
2. Discussion
Inherited CRC is now acknowledged as a significant factor in the development of this disease. Genome sequencing-based diagnosis estimates that one-third of CRC patients appear to have familial colorectal cancer (FCC) as a part of their pathogenesis [39][13]. The initiation of CRC arises from the epithelial tissue due to the accumulation of genetic modulations in specific oncogenes and tumor suppressor genes (TSGs). In the evolution of sporadic CRC, two primary mechanisms of genomic instability have been identified. The first is chromosomal instability (CIN) caused by a cascade of genomic alterations involving activating oncogenes like KRAS and the inactivation of TSG like p53, DCC/SMAD4, and APC. The second, known as microsatellite instability (MSI), is caused by hypermethylation of the promoters of the DNA mismatch repair genes MLH1 and/or MSH2, as well as secondary mutation of genes with coding microsatellites, such as transforming growth factor receptor II (TGF-RII) and BAX [40,41,42][14][15][16]. In comparison, germline mutations in the defined genes may lead to inherited CRC. Those mutations can be in the tumor suppressor gene APC on chromosome 5q as in familial adenomatous polyposis (FAP) or mutated DNA mismatch repair genes in hereditary non-polyposis colorectal cancer (HNPCC) [43,44][17][18]. In this systematic scoping review, differentially expressed candidate proteins in response to γT3 or δT3 treatment were retrieved to identify the molecular mechanisms through which these T3s isoforms modulate anticancer effects. The 16 CBs (BIRC3, BIRC5, CASP3, CASP8, CASP9, CCND1, CDKN1A, CDKN1B, CTNNB1, JUN, MMP9, MYC, PARP1, RELA, VEGFA, and WNT1) short-listed in this study formed three prominent clusters that were part of the apoptotic, transcriptional misregulation, or cancer progression pathways (Figure 2). All three pathways are interlinked and are crucial with respect to anticancer mechanisms. Hence, it is highly likely that the 16 CBs play a pivotal role in mediating anticancer mechanisms induced in human CRC cell lines exposed to γT3 or δT3. A number of the CBs have been reported to play important roles clinically in the carcinogenesis of patients with CRC (Table 1).Table 1. Summary of clinically relevant genes and proteins modulated by tocotrienols in human colon cancer cell lines.
| Pathways Involved | CBs Modulation by T3s | Reported Effects in Colon Cancer Patients | Ref. |
|---|---|---|---|
| Apoptosis | Caspase 3 ( ) ) |
● Irradiated CRC cells from patients with lower levels of caspase-3 was associated with poor prognosis | [45][19] |
| Caspase 8 () |
● Higher prevalence of mutations in the caspase-8 genes in invasive carcinomas; reduce apoptotic activity | [46,47][20][21] | |
| Caspase 9 () |
● Expression of the caspase-9 gene downregulated in CRC tissue compared to surrounding normal mucosa | [48][22] | |
| Transcriptional dysregulation in cancer | CDKN1A (p21) () |
● P21 was downregulated in 50% (371/737) of CRC samples | [49][23] |
Jun family ( ) ) |
● Higher c-Jun expression observed in human colorectal adenocarcinomas | [50][24] | |
| c-MYC () |
● CRC patients with higher MYC expression significantly shorter progression-free survival time and overall survival | [51][25] | |
| RELA (NF-kB/p65) () |
● Reduced RELA expression, resulting in deceased activation of the NF-κB signaling pathway, which inhibited carcinogenesis | [52,53][26][27] | |
| Cancer progression | CCND1 () |
● CCND1 gene was detected in tumors from about 50% (54 out of 111) of CRC patients; absent in normal mucosa | [54][28] |
| VEGFA () |
● Elevated expression of the VEGF family, especially of VEGFA, was reported in CRC patients with LNM | [55][29] | |
| CTNNB1 () |
● CTNNB1 codes for β-catenin, which supports tumor growth ● A significant link between mutations in CTNNB1 gene and MSI |
[56][30] |
Downregulat.ed; Upregulated; CB: candidate biomarkers; CRC: colorectal carcinoma; LNM: lymph node metastasis; MSI: microsatellite instability; T3s: tocotrienols. Italics refer to the gene name of the protein.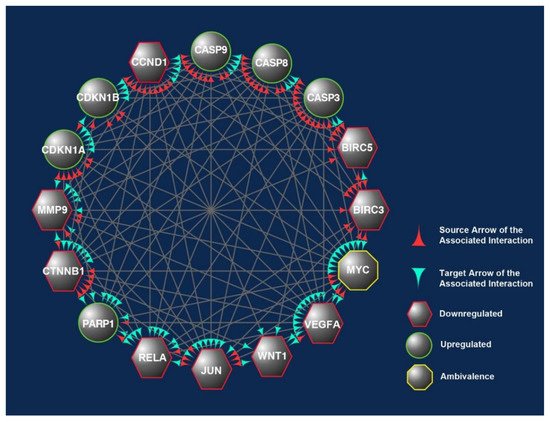
Figure 2. The overlapped targets of the 16 candidate biomarkers (CBs) identified in this study were populated according to their engagements using Cytoscape, an online bioinformatics tool. The protein–protein interactions (PPIs) between these CBs formed a prominent network. The CBs in the hexagons and circles are significantly downregulated or upregulated in human CRC exposed to γT3 or δT3. In contrast, the CB in the octagons is a biomarker that did not show significant changes. The green and red arrowheads reflect the interactions between these CBs. The red arrowheads indicate source CBs protein that can exert stimulatory (circles) or inhibitory (hexagons) or no effects (octagons) on target CBs (green arrowheads).
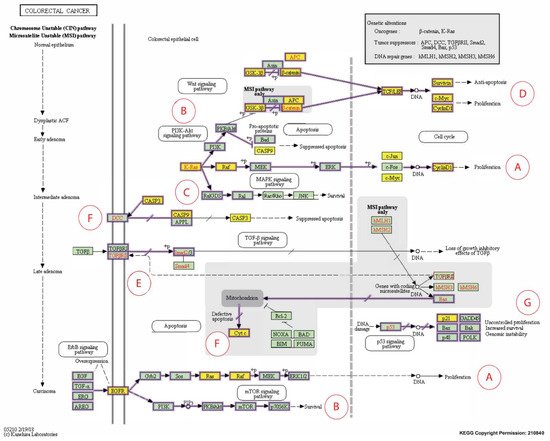

Figure 3. Expanded Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway representation of colorectal carcinoma showing the involvement of the 16 candidate biomarkers selected for this study in the pathogenesis of CRC. The cancer pathways that these 16 CBs are involved in include (A) ERK signaling; (B) PI3K signaling; (C) RAS signaling; (D) WNT signaling; (E) TGFB signaling; (F) Apoptotic signaling; (G) Transcription. Genes marked with yellow rectangles represent the 16 CBs identified in this scoping review.
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386.
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691.
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular mechanisms of action of tocotrienols in cancer: Recent trends and advancements. Int. J. Mol. Sci. 2019, 20, 656.
- Ahn, K.S.; Sethi, G.; Krishnan, K.; Aggarwal, B.B. γ-Tocotrienol inhibits nuclear factor-κb signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J. Biol. Chem. 2007, 282, 809–820.
- Eitsuka, T.; Nakagawa, K.; Miyazawa, T. Down-regulation of telomerase activity in DLD-1 human colorectal adenocarcinoma cells by tocotrienol. Biochem. Biophys. Res. Commun. 2006, 348, 170–175.
- Shibata, A.; Nakagawa, K.; Sookwong, P.; Tsuduki, T.; Tomita, S.; Shirakawa, H.; Komai, M.; Miyazawa, T. Tocotrienol inhibits secretion of angiogenic factors from human colorectal adenocarcinoma cells by suppressing hypoxia-inducible factor-1α. J. Nutr. 2008, 138, 2136–2142.
- Shibata, A.; Nakagawa, K.; Sookwong, P.; Tsuzuki, T.; Oikawa, S.; Miyazawa, T. Tumor anti-angiogenic effect and mechanism of action of δ-tocotrienol. Biochem. Pharmacol. 2008, 76, 330–339.
- Brew, R.; Erikson, J.S.; West, D.C.; Kinsella, A.R.; Slavin, J.; Christmas, S. Interleukin-8 as an autocrine growth factor for human colon carcinoma cells in vitro. Cytokine 2000, 12, 78–85.
- Ananthula, S.; Parajuli, P.; Behery, F.; Alayoubi, A.Y.; El Sayed, K.; Nazzal, S.; Sylvester, P.W. Oxazine derivatives of γ- and δ-tocotrienol display enhanced anticancer activity in vivo. Anticancer. Res. 2014, 34, 2715–2726.
- Xu, W.-L.; Liu, J.-R.; Liu, H.-K.; Qi, G.-Y.; Sun, X.-R.; Sun, W.-G.; Chen, B.-Q. Inhibition of proliferation and induction of apoptosis by γ-tocotrienol in human colon carcinoma HT-29 cells. Nutrition 2009, 25, 555–566.
- Tham, S.-Y.; Loh, H.-S.; Mai, C.-W.; Fu, J.-Y. Tocotrienols modulate a life or death decision in cancers. Int. J. Mol. Sci. 2019, 20, 372.
- Rudner, J.; Jendrossek, V.; Lauber, K.; Daniel, P.T.; Wesselborg, S.; Belka, C. Type I and type II reactions in TRAIL-induced apoptosis–results from dose–response studies. Oncogene 2004, 24, 130–140.
- Burt, R.W. Colon cancer screening. Gastroenterology 2000, 119, 837–853.
- Grady, W.M. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004, 23, 11–27.
- Rajagopalan, H.; Nowak, M.A.; Vogelstein, B.; Lengauer, C. The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer 2003, 3, 695–701.
- Nojadeh, J.N.; Sharif, S.B.; Sakhinia, E. Microsatellite instability in colorectal cancer. EXCLI J. 2018, 17, 159–168.
- Lynch, H.T.; De La Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932.
- Anwar, S.; Hall, C.; White, J.; Deakin, M.; Farrell, W.; Elder, J. Hereditary non-polyposis colorectal cancer: An updated review. Eur. J. Surg. Oncol. (EJSO) 2000, 26, 635–645.
- Zhang, Z.; Wang, M.; Zhou, L.; Feng, X.; Cheng, J.; Yü, Y.; Gong, Y.; Zhu, Y.; Li, C.; Tian, L.; et al. Increased HMGB1 and cleaved caspase-3 stimulate the proliferation of tumor cells and are correlated with the poor prognosis in colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34, 1–9.
- Kim, H.S.; Lee, J.W.; Soung, Y.H.; Park, W.S.; Kim, S.Y.; Park, J.Y.; Cho, Y.G.; Kim, C.J.; Jeong, S.W.; Nam, S.W.; et al. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology 2003, 125, 708–715.
- Duiker, E.W.; Meijer, A.; Van Der Bilt, A.R.M.; Meersma, G.J.; Kooi, N.; Van Der Zee, A.G.J.; De Vries, E.G.; De Jong, S. Drug-induced caspase 8 upregulation sensitises cisplatin-resistant ovarian carcinoma cells to rhTRAIL-induced apoptosis. Br. J. Cancer 2011, 104, 1278–1287.
- Shen, X.-G.; Wang, C.; Li, Y.; Wang, L.; Zhou, B.; Xu, B.; Jiang, X.; Zhou, Z.-G.; Sun, X.-F. Downregulation of caspase-9 is a frequent event in patients with stage II colorectal cancer and correlates with poor clinical outcome. Color. Dis. 2010, 12, 1213–1218.
- Ogino, S.; Kawasaki, T.; Kirkner, G.; Ogawa, A.; Dorfman, I.; Loda, M.; Fuchs, C. Down-regulation of p21 (CDKN1A/CIP1) is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. J. Pathol. 2006, 210, 147–154.
- Wang, H.; Birkenbach, M.; Hart, J. Expression of jun family members in human colorectal adenocarcinoma. Carcinogenesis 2000, 21, 1313–1317.
- Strippoli, A.; Cocomazzi, A.; Basso, M.; Cenci, T.; Ricci, R.; Pierconti, F.; Cassano, A.; Fiorentino, V.; Barone, C.; Bria, E.; et al. c-MYC expression is a possible keystone in the colorectal cancer resistance to EGFR inhibitors. Cancers 2020, 12, 638.
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-κB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334.
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.; Samowitz, W.S.; Wolff, R.K.; Stevens, J.R.; Herrick, J.S. The NF-κB signalling pathway in colorectal cancer: Associations between dysregulated gene and miRNA expression. J. Cancer Res. Clin. Oncol. 2018, 144, 269–283.
- Balcerczak, E.; Pasz-Walczak, G.; Kumor, P.; Panczyk, M.; Kordek, R.; Wierzbicki, R.; Mirowski, M. Cyclin D1 protein and CCND1 gene expression in colorectal cancer. Eur. J. Surg. Oncol. (EJSO) 2005, 31, 721–726.
- Mazeda, I.; Martins, S.F.; Garcia, E.A.; Rodrigues, M.; Longatto, A. VEGF expression in colorectal cancer metastatic lymph nodes: Clinicopathological correlation and prognostic significance. Gastrointest. Disord. 2020, 2, 267–280.
- Mirabelli-Primdahl, L.; Gryfe, R.; Kim, H.; Millar, A.; Luceri, C.; Dale, D.; Holowaty, E.; Bapat, B.; Gallinger, S.; Redston, M. Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res. 1999, 59, 3346–3351.
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 1–15.
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.-Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930.
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462.
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A molecular connection for cell cycle control, adhesion and invasion in tumor and stroma. Cells 2020, 9, 2648.
- Lanza, G.; Ferracin, M.; Gafà, R.; Veronese, A.; Spizzo, R.; Pichiorri, F.; Liu, C.-G.; Calin, G.; Croce, C.M.; Negrini, M. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer 2007, 6, 1–11.
- Kim, M.; Lee, H.E.; Lee, H.S.; Yang, H.-K.; Kim, W.H. Expression of apoptosis-related proteins and its clinical implication in surgically resected gastric carcinoma. Virchows Arch. 2011, 459, 503–510.
- Taylor, R.; Cullen, S.P.; Martin, S. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241.
- Asadi, M.; Shanaehbandi, D.; Kermani, T.A.; Sanaat, Z.; Zafari, V.; Hashemzadeh, S. Expression level of caspase genes in colorectal cancer. Asian Pac. J. Cancer Prev. 2018, 19, 1277–1280.
- Cacina, C.; YaylIm-Eraltan, I.; Arikan, S.; Saglam, E.K.; Zeybek, U.; Isbir, T. Association between CDKN1A Ser31Arg and C20T gene polymorphisms and colorectal cancer risk and prognosis. Vivo 2010, 24, 179–183.
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251.
- Thomas, R.; Weihua, Z. Rethink of EGFR in cancer with its kinase independent function on board. Front. Oncol. 2019, 9, 800.
- Arango, D.; Mariadason, J.M.; Wilson, A.J.; Yang, W.; Corner, G.; Nicholas, C.; Aranes, M.J.; Augenlicht, L.H. c-Myc overexpression sensitises colon cancer cells to camptothecin-induced apoptosis. Br. J. Cancer 2003, 89, 1757–1765.
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151.
- Rochlitz, C.F.; Herrmann, R.; De Kant, E. Overexpression and amplification of c-myc during progression of human colorectal cancer. Oncology 1996, 53, 448–454.
- Khare, V.; Tabassum, S.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. RNA helicase p68 deploys β-catenin in regulating RelA/p65 gene expression: Implications in colon cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–19.
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 2019, 177, 1035–1049.e19.
More