Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Linn Samira Mari Evenseth and Version 2 by Peter Tang.
The γ-aminobutyric acid (GABA) type B receptor (GABAB-R) belongs to class C of the G-protein coupled receptors (GPCRs). Together with the GABAA receptor, the receptor mediates the neurotransmission of GABA, the main inhibitory neurotransmitter in the central nervous system (CNS).
- GABAB receptors
- orthosteric binding site
- allosteric binding site
- structural mechanisms
- drug development
- baclofen
1. Introduction
The metabotropic γ-aminobutyric acid (GABA) type B receptor (GABAB-R) was first described in 1979 by Dr. Norman Bowery and was acknowledged to play an important inhibitory role in neurotransmission [1][2][3][1,2,3]. The receptor was not cloned successfully until 20 years later because of the lack of high-affinity radioligands and, more importantly, due to the unexpected structural features of the receptor [4]. The GABAB-R belongs to class C of G-protein coupled receptors (GPCRs) together with the metabotropic glutamate receptors (mGluRs), calcium-sensing (CaS), taste 1 and orphan receptors [5]. Until recently, only the structure of the extracellular Venus flytrap (VFT) domain hosting the orthosteric binding was known from X-ray crystallography studies. However, recently, four papers were published, describing the cryogenic electron microscopy (cryo-EM) structures of the full-length receptor in several conformations [6][7][6,7]. The paper by Mao et al. [6] describes both a full-length active conformation bound to the agonist baclofen and the positive allosteric modulator (PAM) (R,S)-5,7-di-tert-butyl-3-hydroxy-3-trifluoromethyl-3H-benzofuran-2-one (BHFF) in the presence of the Gi1-protein, and a full-length inactive antagonist (CGP54626)-bound receptor [6].
2. Structure of the GABAB Receptor
The GABAB-R is an obligate heterodimer composed of the GABAB1 and GABAB2 subunits. Each subunit consists of an extracellular VFT domain linked to a heptahelical transmembrane (7TM) domain, and the linker is shorter in sequence and lacks the cysteine residues that are conserved among other class C GPCRs (called the cysteine rich domain (CRD)) (Figure 1) [8][21]. Radioligand binding studies, site-directed mutagenesis studies and the X-ray crystal structures showed that the orthosteric binding site of GABAB-R is located in the VFT of GABAB1, while ligand binding to the GABAB2 VFT have not been observed [9][22]. Binding studies of isolated GABAB2 subunits and studies manipulating the receptor composition showed that the GABAB2 7TM domain is mainly responsible for recruiting G-proteins, in addition to hosting an allosteric binding site [10][11][12][23,24,25]. There is also evidence from biochemical and biophysical studies that the GABAB1 7TM participates in formation of GABAB-R oligomers by interacting with other GABAB1 7TMs in the inactive state [13][14][15][26,27,28]. The receptor was found to be in equilibrium between heterodimers and oligomers by applying SNAP-tag technology [14][27]. The formation of oligomers represents a new level of complexity as the functional consequence can be the transactivation of receptors, which is currently only established for heterodimers [16][29].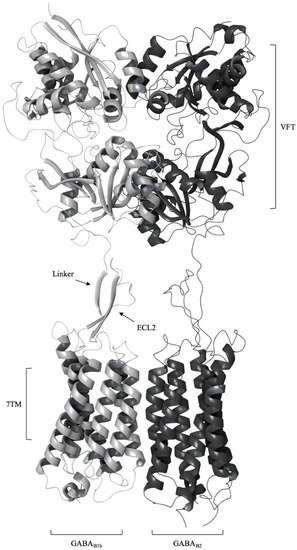
Figure 1. A schematic representation of the (GABA) type B receptor (GABAB-R) heterodimer with the extracellular Venus Flytrap domain (VFT) and the heptahelical transmembrane domain (7TM) of the GABAB1b (gray) and GABAB2 (black) protomers. The VFT is connected to the 7TM by a linker that interacts with the extracellular loop 2 (ECL2) of 7TMs (active receptor conformation (Protein Data Bank (PDB) ID: 6OU8).
3. GABAB Receptor Binding Sites
3.1. Orthosteric Binding Site
The orthosteric binding pocket is located in the crevice of LB1 and LB2 of GABAB1a/b. Binding of agonist induces large conformational changes such that the LB1 and LB2 interact and form a stable closed conformation in timescales necessary for full receptor activation (Figure 2) [23][24][36,50].
Residues located in LB1 are responsible for anchoring both agonists and antagonists in the binding pocket (Figure 2) and their interaction patterns with residues in LB1 are highly similar [23][36]. Mutational studies followed by radioligand- and [35S]GTPγS-binding assays found that mutations of tryptophan (Trp65) and histidine (His170) of GABAB1b abolished antagonist binding [23][36]. Agonist activation was also abolished after mutating Trp65Ala, while the His170Ala mutation only reduced receptor activation [23][36]. Additional mutational studies followed by radioligand binding assays showed that mutating Ser130 to Ala abolished binding of the antagonist CGP54626, while mutation of Ser153 were found to affect the affinity of various ligands differently and are thereby suggested to play a role in selectivity of GABAB ligand recognition [25][51]. For more details about ligand interactions in the orthosteric binding site, please see [23][25][26][27][36,51,52,53].
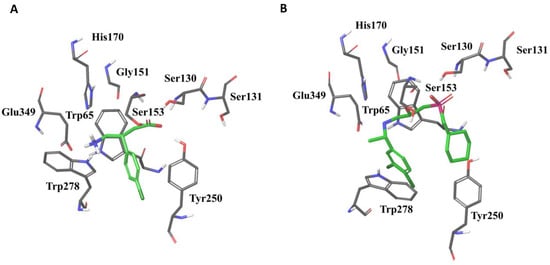
Figure 2. The agonist baclofen (A) and the antagonist CGP54626 (B) in the GABAB1b VFT binding pocket surrounded with residues important for ligand binding (PDB ID: 4MS4 and 4MR7, respectively). Trp278 and Tyr250 are located in Lobe 2 of the GABAB1 VFT, whereas the remaining residues are located in Lobe 1.
Ligand interactions with residues located in LB2 seem to be restricted to agonists and high-affinity antagonists [23][26][27][36,52,53]. Interactions with residues in both LB1 and LB2 are likely to be a requirement for activation, and cause the agonists to become buried within the closed receptor conformation (Figure 2). This is supported by mutational studies indicating that Trp278 and Tyr250 located in LB2 of GABAB1b are critical for agonist binding but have less effect on binding of antagonists [23][28][36,54].
All ligands co-crystalized with the VFT are structural derivatives of GABA with an α-acid and a γ-amino group [23][36]. The α-acid and the γ-amino groups of co-crystalized ligands are stabilized by identical residual interactions in all X-ray crystal structures, independent of intrinsic ligand activity [23][26][27][36,52,53]. Linking receptor interaction patterns to ligand activity and affinity has proven to be difficult, as highly similar compounds show similar receptor interaction patterns despite of different activity [23][36]. Larger and more bulky antagonists, like CGP54626 and CGP62349, are thought to prohibit the formation of a stable closed conformation by forming few and variable interactions with the LB2, likely as a result of the size compared to agonists (Figure 2) [23][26][36,52].
3.2. Allosteric Binding Site
Allosteric modulators change the efficacy and/or affinity of the orthosteric agonist [29][30][55,56]. PAMs potentiate the receptor activation induced by an orthosteric agonist, and some PAMs also display intrinsic agonist activity and are named ago-PAMs [29][55]. FRET studies have also shown that ago-PAMs can actually cause movements of the GABAB1 VFT and the 7TM, corresponding to the conformational changes observed upon agonist activation [31][43]. NAMs inhibit or reduce responses produced by orthosteric agonist, either by stabilizing an inactive conformation of the 7TM domain, and/or decrease the agonist affinity. Currently, less than 100 PAMs are known in the literature to target the GABAB-R, and there is only a single NAM to our knowledge [32][33][57,58]. Silent allosteric modulators (SAMs), also called neutral allosteric ligands (NALs), have no effect on orthosteric agonists efficacy or affinity, but can compete with other allosteric compounds and block their action [29][34][55,59]. Currently, no GABAB receptor SAMs have been described in the literature.
Previous studies have given strong support for an allosteric binding site in 7TM domain of the GABAB2 subunit [35][60]. Binet and coworkers studied various combination of wild type and chimeric GABAB-R subunits and showed that the ago-PAM CGP7930 could activate GABAB2 expressed alone [11][24]. Moreover, investigations of the 7TM domain of GABAB2 by introducing point mutation identified the amino acids Gly706 and Ala708 in TM6 to be important for interactions with PAMs, specifically tested with the PAM GS39783 [36][61]. This study also showed that the PAM GS38783 could bind to a mutated rat GABAB2 subunit and activate the receptor without the GABAB1-subunit present [36][61]. Ligand-guided homology modelling and docking studies gave results in agreement with experimental ligand binding studies (Figure 3), and identified nine amino acids within TM3 (Tyr564 and Leu560), TM5 (Lys664), TM6 (Met702, Cys703, Gly706 and Ser710) and TM7 (Val724 and Cys731) as central for the binding of PAMs to the GABAB2 [32][57]. The binding pocket corresponds to the orthosteric binding pocket in most family A GPCRs. Allosteric modulators have been found to bind within the 7TM of other family C GPCRs as well, as seen for mGlu in multiple studies including mGlu5 crystal structures [37][38][44,62]. Mao et al. describe the cryo-EM structure of the GABAB-R with the ago-PAM BHFF [6], and surprisingly, a novel PAM binding pocket is located in a cavity between TM5–TM6 of GABAB1 and TM6 of GABAB2 [6]. This discovery is supported by Shaye et al., describing two binding sites for the PAM GS39783, site 1 located in the 7TM of GABAB2 and site 2 located at the TM6 heterodimer interface [7]. Mutational studies of these two binding sites showed that site 2 is main allosteric binding site for the PAM GS39783 [7]. Together these studies indicate that there exist at least two allosteric binding sites in the 7TM domains, one within TM3, TM5 and TM6 of the GABAB2 subunit, and another located in the TM6 interface of the 7TMs.
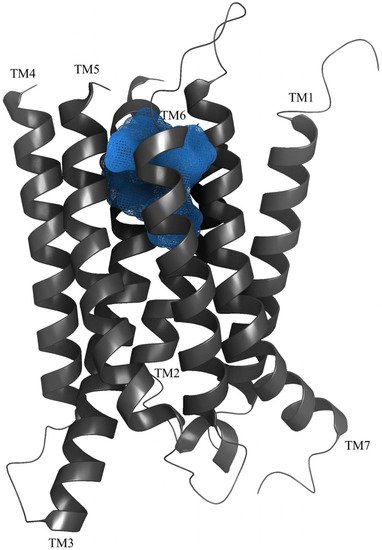
Allosteric compounds are attractive in drug development because of their ability to modulate the effect of orthosteric ligands, and thereby reduce potential side effects and/or increase desirable therapeutic effects [29][55]. Most allosteric modulators exert effects in company with endogenous GABA or other orthosteric ligands [39][63], and when used in combination with an orthosteric drug, lower doses of the orthosteric drug may be required or a specific combination might give beneficial effects due to biased signaling. However, allosteric modulators can also be effective alone, and even in reduced concentrations compared to drugs binding to the orthosteric site [40][64]. Designing allosteric modulators provides the possibility of making compounds that are more selective for the desired functional outcome than orthosteric compounds alone, and thereby potentially reduce the side effects [29][41][55,65]. Distinct ligand signaling profiles are caused by biased signaling, in which different ligands are understood to induce different receptor conformations, allowing potential diverse effector proteins to recognize these conformations. In theory, this can result in a variety of signaling profiles based on the specific allosteric and orthosteric compound in combination or even alone, stabilizing a certain receptor conformation [42][66]. Biased agonism is well characterized for class A GPCRs and class C mGluRs [43][44][67,68], and was recently described for GABAB-R where the PAMs GS39783 and BHF177 were found to have functional selectivity for intracellular signaling pathways in various functional assays [45][69].
4. GABAB Receptor Signaling
The GABAB-R has a complex signaling network, and function as auto- or hetero-receptors on both inhibitory and excitatory nerve terminals. When GABA is released from a GABAergic neuron, it may inhibit further release by binding to presynaptic auto-inhibitory receptors, functioning in a negative feedback loop [46][70]. These auto-receptors can also be activated by GABA released by a single action potential [17][30]. GABAB-Rs are also found on non-GABAergic neurons where they act as hetero-receptors and inhibit the release of other neurotransmitters such as glutamate from glutamatergic neurons [17][30].
Activation of pre- and postsynaptic GABAB-Rs by an agonist results in the inhibition of adenylyl cyclase (AC) through the Gαi/o pathway [19][32]. In presynaptic terminals, binding of Gαi/o to AC causes decreased levels of cAMP, which prevents vesicle fusion and thereby neurotransmitter release [19][32]. In addition, the Gβγ subunit of the G-protein binds directly to voltage-gated Ca2+ channels (VGCC), resulting in inhibition of inward rectifying Ca2+ channels necessary for vesicle fusion [19][32]. The Gβγ subunit can also directly attach to SNAP receptors (SNARE) that are responsible for anchoring vesicles to the synaptic membrane and thereby inhibiting presynaptic membrane vesicle fusion [19][32]. In the postsynaptic membrane, the Gβγ subunit also binds to and inhibits the VGCC, but contributes to a hyperpolarization and inhibits the release of many neurotransmitters including noradrenaline, serotonin and dopamine [19][32]. Postsynaptically, the cAMP-dependent protein kinase A (PKA) signaling pathway is affected by the inhibition of AC [19][32], resulting in inhibition or reduced permeability of ion channels such as the ionotropic glutamate NMDA receptor that mediates Ca2+ influx [47][71]. In addition, the Gβγ subunit stimulates the G-protein coupled inwardly rectifying K+ channels (GIRK), resulting in inhibition of the postsynaptic potential and decreased long-term potentiation (LTP) [48][49][72,73]. The phosphorylation of the Extracellular Signal-Regulated Protein Kinases 1 and 2 (ERK1/2) in certain areas of the hippocampus, known to be important for memory and learning, has also been linked to GABAB-R activation [50][74]. ERK1/2 play an important role in gene expression by regulating the activity of transcription factors. A study showed that GABA and baclofen can increase the phosphorylation of ERK1/2 without changing the expression level in cerebellar granule neurons cultured from mouse [50][74]. The phosphorylation was found to be G-protein dependent as the pertussis toxin known to inhibit Gi/o-protein coupling, also inhibited phosphorylation of ERK1/2 [50][74]. The activation of the receptor has also been linked to direct interactions with the L-type VGCC isoforms CaV1.2 and CaV1.3, which increase channel activity and mediate ERK1/2 phosphorylation via these interactions [51][75]. These ion channels contain multiple consensus sites for phosphorylation by protein kinases, and both phospholipase C (PLC) and protein kinase C (PKC) are suggested to be involved in GABAB-R-mediated facilitation of these channels [51][75]. The link between ERK1/2 and GABAB-Rs is as described, highly complex and additional efforts are needed to clarify the full aspect of pathway-specific activation and functional selectivity that is also likely to be cell type specific.
The C-terminal GABAB-R region also serves as binding site for multiple proteins, including regulatory G-protein signaling (RGS) proteins that regulate receptor activity. In addition, leucine-zipper transcription factors, scaffolding and adaptor proteins interact with the coiled-coil C-terminus of the receptor and modulate intracellular trafficking, receptor dimerization and synaptic localization contributing to the functional diversities of the GABAB-R [52][76]. GABAB-R signaling is also regulated by the auxiliary protein subunits, KCTDs, which control the kinetics of GIRK activation and desensitization. These effects are mediated by the binding of the KTCDs to the C-terminal of the GABAB2 subunit and to the Gβγ proteins [22][35].
5. GABAB Receptor Pathophysiology
The disruption of GABAB-R signaling pathways is linked to a variety of neuropsychiatric disorders and diseases including depression, anxiety, schizophrenia, addiction, learning and memory, epilepsy, neurodegenerative disorders, cancer and gastroesophageal reflux disorder (GERD) [53][54][55][56][57][58][12,18,77,78,79,80]. The autoimmune disease Anti-GABAB-R encephalitis was recently described in a case report and comprises a new category of GABAB-R related diseases, where patients develop antibodies against the receptor [58][59][80,81]. Recent evidence suggests that neurotransmitters are involved in tumor development and proliferation of multiple cancer types [60][61][11,82]. The GABAB-R is found to be upregulated in a variety of cancer cell lines including hepatocellular and colon cancer cells [61][82]. Immunohistostaining of tumor samples from the thyroid gland found a significantly increase in GABAB2 expression in tumor tissue compared to normal tissue and linked the expression to malignancy [61][82], while increased expression of GABAB1 has been linked to malignancy in human breast cancer [61][82]. The role of GABAB-R activation for cell proliferation has been investigated by baclofen administration in different rat cancer models, such as gastric cancer and colon tumor models. The results showed that baclofen reduced the incident of gastric cancer significantly, and decreased the colon tumor malignancy [61][82]. Additional in vitro studies have displayed an inhibitory role of baclofen in tumor cell proliferation and/or migration in numerous cell lines including human pulmonary adenocarcinoma-, pancreatic duct epithelial and small airway epithelial cells. However, certain prostate cancer cell lines where not inhibited after baclofen treatment, and rather enhanced cancer migration probably by promoting matrix metalloproteinase-3 production [61][82].
Epilepsy is a disease caused by abnormal neural activity and is characterized by seizures [62][83]. There are different types of seizures depending on the neuronal network involved, and the role of GABAB-R depends on the type of seizure and network involved [62][83]. The GABAB-R-mediated mechanisms represent an dichotomy, as agonists might exacerbate some type of seizures and act as an anticonvulsant in other types [62][83]. Antagonists have also shown anticonvulsant properties in certain seizure types. The complex pathology of epilepsy, the adverse effects of GABAB-R agonists and the lack of appropriate and approved antagonists, indicate that treatment of this disease by targeting the GABAB-R must await clinical development of new ligands.
The stimulation of GABAB-R by baclofen is linked to a reduction in addiction-related behavior towards substances such as nicotine, cocaine and alcohol in animal models [57][63][79,84]. Drugs of abuse stimulate the mesolimbic system in the brain that controls the release of the reward-associated neurotransmitter dopamine [64][85]. A recent study showed that the rewarding effect of nicotine was reduced in animals pre-treated with baclofen [64][85]. Nicotine stimulates the nicotine acetylcholine receptors (nAchRs) located on GABAergic, glutamatergic and dopaminergic neurons, which causes release of dopamine. Baclofen activates GABAB-Rs in dopaminergic and GABAergic neurons, and significantly reduces the amount of dopamine release by inhibition of the dopaminergic neurons.
Baclofen and PAMs have also demonstrated anxiolytic effects in animal cognition models and have been implicated to reverse anxiogenic responses from addiction-related withdrawal [54][65][66][67][18,86,87,88]. Shortly after the first published description of the GABAB-R, the receptor was linked to depression [68][69][89,90], and GABAB-R antagonists have shown to exhibit antidepressant effects in a variety of animal models [54][65][70][18,86,91]. Abnormal peripheral serum concentrations of GABA and glutamate, and reduced brain levels of the enzyme glutamic acid decarboxylase (GAD), which is responsible for converting glutamate to GABA, have been found in young adults diagnosed with depression and schizophrenia [55][77]. Changes in GABAergic neurons and in the concentration of GABAB-R isoforms, have been discovered in post-mortem examination of patients diagnosed with clinical depression, and support the theory of involvement of the GABAergic system in psychiatric disorders [55][71][72][77,92,93].
PAMs and NAMs are under investigation for treatment of neuropsychiatric disorders [73][10], but currently no allosteric modulator is marketed for therapeutic use.
6. The GABAB Receptor in Drug Discovery
In 1967, GABA was recognized as the main inhibitory neurotransmitter in the mammalian CNS though the presence of the neurotransmitter was recognized nearly 20 years earlier [74][75][94,95]. The GABA molecule has been known for several years and was synthesized already in 1888 [74][75][94,95]. GABA is a small neurotransmitter with the molecular weight of 103 Da, has high hydrophilicity and high aqueous solubility and cannot penetrate the BBB (Figure 4). Various drug discovery and development regimes have been implemented during the last decades to find drug-like GABA analogues with more appropriate physicochemical properties for possible therapeutic application. These efforts have resulted in many GABA analogues, but, surprisingly, many of these do not bind the receptor, despite high structural similarity to GABA [76][96]. This emphasizes the apparent struggle of developing selective and potent ligands, but also highlight the urge for new ligands with chemotypes different from present GABAB-R compounds. New ligands may enhance our understanding of activation mechanisms, clarify the role of the receptor in different signaling pathways and potentially benefit in the understanding of therapeutic effects or become new drugs.
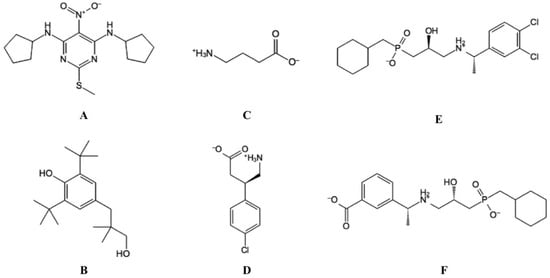
Figure 4. The structure of selected ligands targeting the GABAB-R. (A) GS39783, (PAM). (B) CGP7930, (ago-PAM). (C) GABA (endogenous agonist). (D) Baclofen (agonist), and (E) CGP54626 (antagonist). (F) CGP56999A (antagonist).