The Multiple sclerosis (MS) is the most common autoimmune and demyelinating disease of the central nervous system (CNS), characterized, in the majority of cases, by initial relapses that later evolve into progressive neurodegeneration, severely impacting patients’ motor and cognitive functions. Despite the availability of immunomodulatory therapies effective to reduce relapse rate and slow disease progression, they all failed to restore CNS present manuscript provides a comprehensive review of microglial myelin that is necessary for MS full recovery. Microglia are the primary inflammatory cells present in MS lesions, therefore strongly contributing to demyelination and lesion extension. Thus, many microglial-based therapeutic strategies have been focused on the suppression of microglial pro-inflammatory phenotype and neurodegenerative state to reduce disease severityphagocytosis and its involvement in MS development and repair.
- Microglia
- Phagocytosis
- Neurodevelompent
- Demyelination
- Remyelination
- Multiple Sclerosis
1. InAbstroduaction
Microglia, the resident ultiple sclerosis (MS) is the most common autoimmune cells and macrophagesand demyelinating disease of the CNS, make up only 15% of the total brain cellsentral Nervous System (CNS), characterized, in the [1] mandjority 10% of whole glial cells [2][3]. Nonethof caseless, due to their essential functions as immune mediators and primary phagocytes of the brain and spinal cord, microglia have attracted much attention and research. This yolk sac-derived population colonizes the brain during embryogenesis and differentiates under the influence of CNS microenvironment, by initial relapses that later evolve into progressive neurodegeneration, severely impacting patients’ motor and cognitive functions. [4], rDespiding in the healthy nervous system as a highly stable population with long and ramified processes interacting with blood vessels, neurons, and other glial cells in a dynamic “surveillant state”te the availability of immunomodulatory therapies effective to reduce relapse rate [5]. “Surveillant”d microglia maintain CNS homeostasis by sensing pathologic events, scavenging pathogen-associated molecular patterns (PAMPs)/danger-associated molecular patterns (DAMPs), and by phagocytosing dead cells and misfolded proteinsslow disease progression, they all failed to restore CNS myelin -necessary for MS full recovery. Microglia are [6][7]. At the same time, in physiological conditions, they are also engaged in the regulation of biological processes either remodeling synaptic plasticity [8][9], suppprimary inflammatory cells present in MS lesions, therefore strorting neuronal survival [10][33], ly contribrain sexual differentiationting [11][12], tor promoting proper demyelination [13][14][15].
2. Microglial Phagocytosis
New compouands, in non-clinical testing, revealed some encouraging effects regardinglesion extension. Thus, many microglial phagocytosis (Figure 1). To b-basegind with, studies from Chen et al. found that the incubattherapeutic strategies have been focused on the suppression of primary mmicroglia with two omega-3 polyunsaturated fatty acids-docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA)—modulated the cell l pro-inflammatory phenotype towards a more rand neurodegenerative state and enhanced myelin phagocytosis in vitro. Attractively, such treatment in vivo reduced demyelination and ameliorated motor and cognitive functions in a cuprizone deto reduce disease severity. On the other hand, the contribution of myelinating model [16]. Additi phagocytosis advonally encouraging, a human recombinant IgM antibody (rHIgM22) effectively modulatedating the neuroprotective role of microglial myelin phagocytosis. Indeed, in vitro testing showed that rHIgM22 could bind to myelin, tagging debris for their internalization by microglia, in a complement-mediated manner [17], and, when admini in MS has been less explored. Indeed, despite the presence of functional oligodendrocyte precurstrated in vivo, rHIgM22 promoted OPC differentiation, accelerated remyelination, and even ameliorated memory deficits in rodent models of cuprizone and chronic virus-induced der cells (OPCs), within lesioned areas, MS plaques fail to remyelination [18][19][20]. Giving this newly association between phagocytosis and microglial neuroprotective functions over brain pathologies, two more studies concerning mods a result of the over-accumulation of microglial phagocytosis have just been published. In the first one, the authors treated ratyelin-toxic debris that must be cleared away by microglial cells with endocannabinoid 2-AG. By enhancing the mRNA levels of phagocytosis associated genes (cd206, sirp1α, msr1, and Trem2), endocannabinoid 2-AG boosted phagocytosis of both Coli-coated beads and rat-purified myelin debris. Moreover, using a mice model of MS, the Theiler’s induced d. Dysregulation of this process has been associated with the impaired neuronal recovery and deficient remyelinating disease model (TMEV-IDD), TMEV-induced mice treated with endocannabinoid 2-AG showed augmented microglial myelin phagocytosis in the corpus callosum that likely resulted from the observed upregulation of msr1 and Lamp1 mRNA levels, genes associated with mion. In line with this, here we provide a comprehensive review of microglial myelin phagocytic machinery and phagosome formation, respectively. Interestingly, these animals had increased OPC differentiationosis and its involvement in MS development and remyelination following demyelination in the corpus callosumpair. Alongside, we discuss [21]. In the spotecond study, Liu et al. observed that a new pharmacological treatment with pseudoginsenoside-F11 accelerated CR3-dependent myelin phagocytosis in a microglial culture after oxygen-glucose deprivntial of phagocytic-mediated therapeutic approaches and encourage their modulation (OGD) and permanent middle cerebral artery occlusion (pMCAO) in vivo and, by doing so, decreased de infarct area and improved neurological functions in pMCAO-treated rats [22]as a novel and rational approach to ameliorate MS-associated pathology.
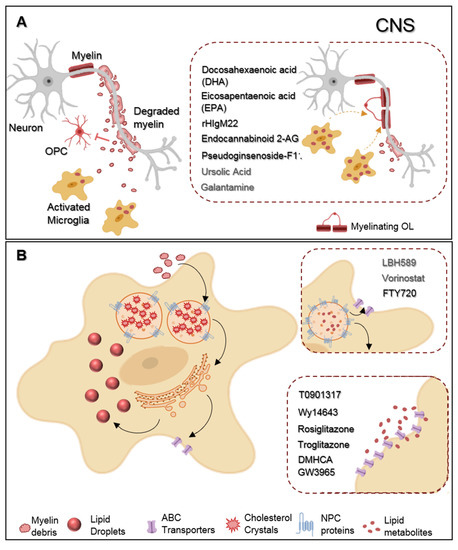
Figure 1.
A
B