Preeclampsia (PE) is a specific syndrome of human pregnancy, being one of the main causes of maternal death. Persistent inflammation in the endothelium stimulates the secretion of several inflammatory mediators, activating different signaling patterns. One of these mechanisms is related to NLRP3 activation, initiated by high levels of danger signals such as cholesterol, urate, and glucose, producing IL-1, IL-18, and cell death by pyroptosis. Furthermore, reactive oxygen species (ROS), act as an intermediate to activate NLRP3, contributing to subsequent inflammatory cascades and cell damage. Moreover, increased production of ROS may elevate nitric oxide (NO) catabolism and consequently decrease NO bioavailability. NO has many roles in immune responses, including the regulation of signaling cascades. At the site of inflammation, vascular endothelium is crucial in the regulation of systemic inflammation with important implications for homeostasis.
- NLRP3
- preeclampsia
- nitric oxide
- reactive oxygen species
1. Introduction
2. Preeclampsia and Endothelial Dysfunction
Endothelial dysfunction is the term used to describe an imbalance in these endothelial functions affecting vasoprotective homeostasis [20][14].
In normal pregnancies, the typical increase in blood volume is commonly compensated by a slight decrease in blood pressure. For a long time, this rise in blood pressure has been associated with reduced maternal vascular resistance [21,22][15][16]. However, in PE, the compensatory maternal vascular adaptations are insufficient, and it has been associated with systemic endothelial dysfunction [23,24,25,26][17][18][19][20]. In this syndrome, this characteristic is associated with the PE poorly perfused placenta, which releases proinflammatory and antiangiogenetic factors into maternal circulation [27,28][21][22]. This hypothesis has been reinforced by many studies so far. For example, Myers and colleagues demonstrated that healthy myometrium vessels incubated with plasma from PE pregnant women had reduced endothelium mediated vascular relaxation compared to those incubated with plasma from healthy pregnant women [29][23]. Plasma from women with PE can modify the endothelial function, altering the balance between vasoactive substances. Despite evidence showing the release of placental factors into maternal circulation could alter endothelial function, the exact mechanism is not fully understood. [30][24].
Until today, many studies demonstrated several alterations, both locally and in circulation, in multiple bioactive factors in PE. For example, the angiogenic balance disturbance has been described by decreases in pro-angiogenic vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) by the action of the placental soluble fms-like tyrosine kinase-1 (sFlt-1) and soluble endoglin (sEng) [29,31,32,33][23][25][26][27]. Moreover, proinflammatory molecules such as tumor necrosis factor-α (TNF-α), endocan, interleukin-6 (IL-6), and IL-1β have also been reported to be altered in PE [34,35,36,37][28][29][30][31]. Altogether, these alterations lead to systemic endothelial dysfunction in PE, and it also is possible that there may be more mechanisms involved that were not discovered yet.
3. NLRP3 Inflammasome Activation and Regulation in Preeclampsia
3.1. Inflammasome Formation and the Role of NLRP3 in the Pathogenesis of PE
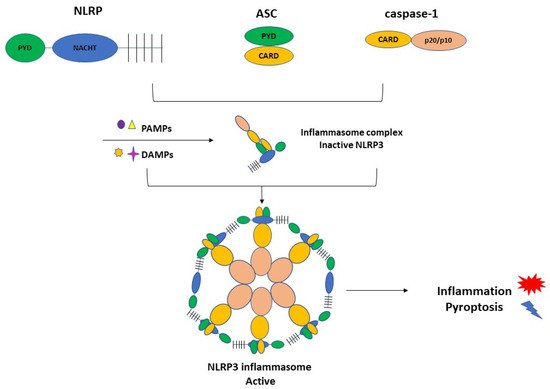
3.2. Activation of NLRP3 and Pyroptosis: The Cell Death Related to Inflammatory Processes
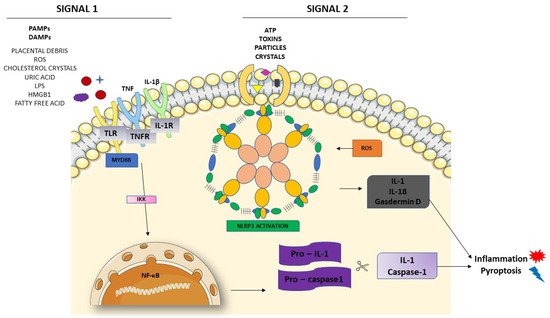
4. NLRP3 and its Relation with Endothelial Dysfunction and Oxidative Stress
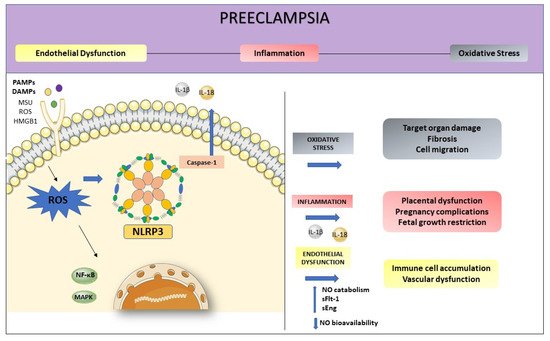
5. Conclusions
References
- American College of Obstetricians and Gynecologists. ACOG practice bulletin number 222. Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 2020, 135, 237–260.
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis, and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens. 2014, 4, 97–104.
- Mol, B.W.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; de Groot, C.J.; Hofmeyr, G.J. Pre-eclampsia. Lancet 2016, 387, 999–1011.
- Kintiraki, E.; Papakatsika, S.; Kotronis, G.; Goulis, D.G.; Kotsis, V. Pregnancy-Induced hypertension. Horm 2015, 14, 211–223.
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815.
- Opitz, B.; Hippenstiel, S.; Eitel, J.; Suttorp, N. Extra- and intracellular innate immune recognition in endothelial cells. Thromb. Haemost. 2007, 98, 319–326.
- Li, Y.X.; Wang, P.; Yang, X.; Wang, W.; Zhang, J.; He, Y.; Zhang, W.; Jing, T.; Wang, B.; Lin, R. SIRT1 inhibits inflammatory response partly through regulation of NLRP3 inflammasome in vascular endothelial cells. Mol. Immunol. 2016, 77, 148–156.
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.L.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: Role of HMGB1. J. Cell Mol. Med. 2015, 19, 2715–2727.
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016, 94, 1335–1347.
- Zhang, Y.; Li, X.; Pitzer, A.L.; Chen, Y.; Wang, L.; Li, P.L. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: Beyond inflammation. Antioxid. Redox Signal 2015, 22, 1084–1096.
- Weel, I.C.; Romão-Veiga, M.; Matias, M.L.; Fioratti, E.G.; Peraçoli, J.C.; Borges, V.T.; Araujo Jr, J.P.; Peraçoli, M.T. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J. Reprod. Immunol. 2017, 123, 40–47.
- Xu, L.; Li, S.; Liu, Z.; Jiang, S.; Wang, J.; Guo, M.; Zhao, X.; Song, W.; Liu, S. The NLRP3 rs10754558 polymorphism is a risk factor for preeclampsia in a Chinese Han population. J. Matern. Fetal Neonatal Med. 2019, 32, 1792–1799.
- Pontillo, A.; Reis, E.C.; Bricher, P.N.; Vianna, P.; Diniz, S.; Fernandes, K.S.; Chies, J.A.; Sandrim, V. NLRP1 L155H Polymorphism is a Risk Factor for Preeclampsia Development. Am. J. Reprod. Immunol. 2015, 73, 577–581.
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: From mechanism to pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967.
- Chesley, L.C.; Talledo, E.; Bohler, C.S.; Zuspan, F.P. Vascular reactivity to angiotensin II and norepinephrine in pregnant and nonpregnant women. Am. J. Obstet. Gynecol. 1965, 91, 837–842.
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.; Menheere, P.P.; Peeters, L.H. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am. J. Obstet. Gynecol. 1993, 169, 382–392.
- Amaral, L.M.; Wallace, K.; Owens, M.; LaMarca, B. Pathophysiology and Current Clinical Management of Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 19–21.
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112.
- Redman, C.W.G. Pre-eclampsia and the placenta. Placenta 1991, 12, 301–308.
- Yoshida, A.; Nakao, S.; Kobayashi, M.; Kobayashi, H. Flow-mediated vasodilation and plasma fibronectin levels in preeclampsia. Hypertension 2000, 36, 400–404.
- Maynard, S.E.; Min, J.; Merchan, J.; Lim, K.; Li, J.; Mondal, S. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Rinehart, B.K.; Terrone, D.A.; Lagoo-Deenadayalan, S.; Barber, W.H.; Hale, E.A.; Martin, J.N., Jr.; Bennett, W.A. Expression of the placental cytokines tumor necrosis factor-alpha, interleukin 1beta, and interleukin 10 is increased in preeclampsia. Am. J. Obstet. Gynecol. 1999, 181, 915–920.
- Myers, J.; Mires, G.; Macleod, M.; Baker, P. In preeclampsia, the circulating factors capable of altering in vitro endothelial function precede clinical disease. Hypertension 2005, 45, 258–263.
- Roberts, J. Endothelial Dysfunction in Preeclampsia. Semin. Reprod. Endocrinol. 1998, 16, 5–15.
- Akhilesh, M.; Mahalingam, V.; Nalliah, S.; Ali, R.M.; Ganesalingam, M.; Haleagrahara, N. Hypoxia-inducible factor-1α as a predictive marker in pre-eclampsia. Biomed. Rep. 2013, 1, 257–258.
- Rana, S.; Karumanchi, S.A.; Levine, R.J.; Venkatesha, S.; Rauh-Hain, J.A.; Tamez, H.; Thadhani, R. Sequential changes in antiangiogenic factors in early pregnancy and risk of developing preeclampsia. Hypertension 2007, 50, 137–142.
- Romero, R.; Nien, J.K.; Espinoza, J.; Todem, D.; Fu, W.; Chung, W.; Kusanovic, J.P.; Gotsch, F.; Erez, O.; Mazaki-Tovi, S.; et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J. Matern. Fetal Neonatal Med. 2008, 21, 9–23.
- Cakmak, M.; Yilmaz, H.; Bağlar, E.; Darcin, T.; Inan, O.; Aktas, A.; Celik, H.T.; Ozdemir, O.; Atalay, C.R.; Akcay, A. Serum levels of endocan correlate with the presence and severity of pre-eclampsia. Clin. Exp. Hypertens. 2016, 38, 137–142.
- Pinheiro, M.B.; Gomes, K.B.; Ronda, C.R.S.C.; Guimarães, G.G.; Freitas, L.G.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; Dusse, L.M. Severe preeclampsia: Association of genes polymorphisms and maternal cytokines production in Brazilian population. Cytokine 2014, 71, 232–237.
- Ramma, W.; Buhimschi, I.A.; Zhao, G.; Dulay, A.T.; Nayeri, U.A.; Buhimschi, C.S.; Ahmed, A. The elevation in circulating anti-angiogenic factors is independent of markers of neutrophil activation in preeclampsia. Angiogenesis 2012, 15, 333–340.
- Siljee, J.E.; Wortelboer, E.J.; Koster, M.P.H.; Imholz, S.; Rodenburg, W.; Visser, G.H.A.; de Vries, A.; Schielen, P.C.J.I.; Pennings, J.L.A. Identification of interleukin-1 beta, but no other inflammatory proteins, as an early onset pre-eclampsia biomarker in first-trimester serum by bead-based multiplexed immunoassays. Prenat. Diagn. 2013, 33, 1183–1188.
- Socha, M.W.; Malinowski, B.; Puk, O.; Dubiel, M.; Wiciński, M. The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia. Cells 2020, 9, 1642.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Xie, F.; Hu, Y.; Turvey, S.E.; Magee, L.A.; Brunham, R.M.; Choi, K.C.; Krajden, M.; Leung, P.C.K.; Money, D.M.; Patrick, D.M.; et al. Toll-like receptors 2 and 4 and the cryopyrin inflammasome in normal pregnancy and pre-eclampsia. BJOG 2010, 117, 99–108.
- Matias, M.L.; Romão, M.; Weel, I.C.; Ribeiro, V.R.; Nunes, P.R.; Borges, V.T.M.; Araújo, J.P., Jr.; Peraçoli., J.C.; de Oliveira, L.; Peraçoli, M.T. Endogenous and uric acid-induced activation of NLRP3 inflammasome in pregnant women with preeclampsia. PLoS ONE 2015, 10, e0129095.
- Mulla, M.J.; Myrtolli, K.; Potter, J.; Boeras, C.; Kavathas, P.B.; Sfakianaki, A.K.; Tadesse, S.; Norwitz, E.R.; Guller, S.; Abrahams, V.M. Uric acid induces trophoblast IL-1beta production via the inflammasome: Implications for the pathogenesis of preeclampsia. Am. J. Reprod. Immunol. 2011, 65, 542–548.
- Shirasuna, K.; Usui, F.; Karasawa, T.; Kimura, H.; Kawashima, A.; Mizukami, H.; Ohkuchi, A.; Nishimura, S.; Sagara, J.; Noda, T.; et al. Nanosilica-induced placental inflammation and pregnancy complications: Different roles of the inflammasome components NLRP3 and ASC. Nanotoxicology 2015, 9, 554–567.
- Tamura, K.; Ishikawa, G.; Yoshie, M.; Ohneda, W.; Nakai, A.; Takeshita, T.; Tachikawa, E. Glibenclamide inhibits NLRP3 inflammasome-mediated IL-1beta secretion in human trophoblasts. J. Pharmacol. Sci. 2017, 135, 89–95.
- Brien, M.E.; Duval, J.C.; Palacios, J.; Boufaied, I.; Hudon-Thibeault, A.-A.; Nadeau-Valle’e, M.; Vaillancourt, C.; Sibley, C.P.; Abrahams, V.M.; Jones, R.L.; et al. Uric acid crystals induce placental inflammation and alter trophoblast function via an IL-1-dependent pathway: Implications for fetal growth restriction. J. Immunol. 2017, 198, 443–451.
- Shirasuna, K.; Karasawa, F.T.; Usui, M.; Kobayashi, T.; Komada, H.; Kimura, A.; Kawashima, A.; Ohkuchi, A.; Taniguchi, S.; Takahashi, M. NLRP3 deficiency improves angiotensin II-induced hypertension but not fetal growth restriction during pregnancy. Endocrinology 2015, 156, 4281–4292.
- Seno, K.; Sase, S.; Ozeki, A.; Takahashi, H.; Ohkuchi, A.; Suzuki, H.; Matsubara, S.; Iwata, H.; Kuwayama, T.; Shirasuna, K. Advanced glycation end products regulate interleukin-1b production in human placenta. J. Reprod. Dev. 2017, 63, 401–408.
- Gomez-Lopez, N.; Motomura, K.; Miller, D.; Garcia-Flores, V.; Galaz, J.; Romero, R. Inflammasomes: Their Role in Normal and Complicated Pregnancies. J. Immunol. 2019, 203, 2757–2769.
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95.
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2017, 113, 5.
- Chen, Z.; Martin, M.; Li, Z.; Shyy, J.Y. Endothelial dysfunction: The role of sterol regulatory element-binding protein-induced NOD-like receptor Family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr. Opin. Lipido. 2014, 25, 339–349.
- Wang, J.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL- 1beta-dependent manner. Blood 2011, 118, 2366–2374.
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018, 122, 1722–1740.
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal 2015, 22, 1111–1129.
- Dunn, D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species, and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Shihata, W.A.; Michell, D.L.; Andrews, K.L.; Chin-Dusting, J.P. Caveolae: A role in endothelial inflammation and mechanotransduction? Front. Physiol. 2016, 7, 628.