Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Mariarosa Anna Beatrice Melone.
Neurodegenerative diseases (NDs) represent a heterogeneous group of aging-related disorders featured by progressive impairment of motor and/or cognitive functions, often accompanied by psychiatric disorders. NDs are denoted as ‘protein misfolding’ diseases or proteinopathies, and are classified according to their known genetic mechanisms and/or the main protein involved in disease onset and progression.
- neurodegenerative diseases
- blood–brain barrier
- brain targeting
- nanoformulations
1. Introduction: Neurodegenerative Diseases
In recent years, great advances have been made in understanding the genetic, molecular, and biochemical mechanisms of neurodegenerative diseases (NDs), a group of disorders of the central nervous system (CNS) featured by extra- and intra-cellular accumulation of misfolded proteins, as well as progressive dysfunction, degradation or death of neurons. However, surviving neurons show remarkable morphological changes in the size and shape of the nucleus and chromatin condensation [1].
Protein misfolding is the main cause of a series of strictly connected events, first and foremost, the failure of the ubiquitin-proteasome system (UPS), followed by the collapse of autophagy. Both events lead to oxidative stress, mitochondrial energy deficiency, dysfunction of neurotrophins, and activation of neuroinflammation, as well as extensive formation of free radicals. Concomitantly, dysfunctions of the neuronal Golgi apparatus and axonal transport can frequently occur [1].
Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) are considered worldwide as the main neurodegenerative disorders [2][3], sharing several similarities at the subcellular and molecular level such as synaptic abnormalities, deposition of misfolded proteins in the brain, activation of glial cells, and loss of neuronal connectivity of synaptic circuits. All these diseases usually appear in adulthood as slowly progressive disorders, mainly affecting motor, cognitive, and mental functions [4][5].
1.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is the most prevalent cause of dementia, with the number of patients expected to reach more than 150 millions by mid-century worldwide. AD is a slowly progressive ND, which gradually impairs activities of daily living and social functioning in affected people [2][6][7][8].
The development of AD symptoms is very insidious: it takes many years before patients begin to show memory impairment exceeding that typically observed for their age group and many more years before their cognitive abilities decline to a functionally disabling degree, with loss of spatial and temporal orientation, as well as verbal fluency [9][10].
In most cases, AD is late-onset and occurs sporadically, but some early-onset familial forms have been described, mainly linked to three causative genes, i.e. APP, PSNE1, and PSEN2, which respectively code amyloid precursor protein (APP), presenilin-1 (PSNE1), and presenilin-2 (PSNE2) proteins. In addition, several genetic risk factors, including the ε4 allele of apolipoprotein E (ApoE), have been reported [11].
Currently, there is no definitive treatment for AD, and the available therapeutic interventions (Figure 1) are aimed at managing the disease, i.e., mitigating or halting the associated symptoms [12][13][14][15].
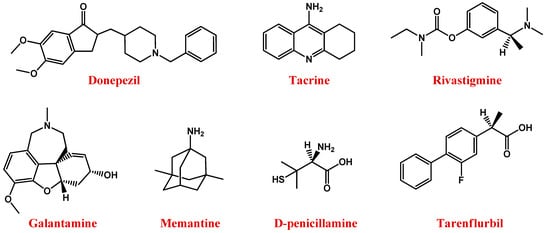
Figure 1. Molecular structures of main anti-AD drugs.
In sporadic AD, a reduction in brain weight, although not constant, is generally usual, as evidenced by macroscopic findings. More severe atrophy is evident in early-onset and familial AD. Diffuse gyral atrophy and ventricular dilatation—involving the temporal cortex, amygdala, hippocampus, and entorhinal cortex, without affecting the occipital lobe—are often found.
Microscopic neuropathological features of AD—detected on post-mortem brain examination—include intracellular neurofibrillary tangles (NFTs), and extracellular plaques of β-amyloid peptide (Aβ), both associated with neuronal loss and altered synaptic connectivity [16][17][18][19].
Under physiological conditions, microtubule-associated tau protein supports neuronal growth, but becomes cytotoxic when hyperphosphorylated, precipitating as paired helical filaments, i.e., NFTs. Tau aggregation affects neuronal axons and, consequently, causes neurodegeneration, with significant effects on AD pathogenesis and progression [20][21][22][23][24].
In turn, according to the Aβ cascade hypothesis, the formation of Aβ plaques is essentially generated from the catalytic cleavage of APP, a transmembrane glycoprotein composed of 770 amino acids, operated first by β-secretase and then by γ-secretase, respectively, at the N- and C-termini of the protein [25][26][27].
The deposition of the cleavage product of APP is ultimately the strong self-aggregating β-amyloid peptide of 42 amino acids, known as Aβ1–42 [28], which is neurotoxic both in vitro and in vivo [29]. Aβ monomers form dimers, then oligomers, protofibrils, and mature fibrils, finally resulting in the formation of Aβ aggregates [30].
Accumulation of Aβ1–42 promotes further pathological effects, such as disruption of synaptic connections with consequent neuronal damage and decreased release of cholinergic neurotransmitters. Indeed, the cholinergic system plays an important role in learning so that cholinergic dysfunctions are the main causes of memory impairments observed in AD patients [31][32]. Therefore, many treatment strategies for AD have been focused on the restoration of the cholinergic neurotransmission [33][34].
In this context, available drugs are mainly cholinesterase inhibitors (ChEIs)—including donepezil [35], tacrine [36], rivastigmine [37], and galantamine [38] (Figure 1) —which prevent acetylcholine breakdown, improving its bioavailability [33][34].
Memantine (Figure 1), an uncompetitive antagonist of glutamatergic N-methyl-d-aspartate (NMDA) receptors, represents a valuable therapeutic option for AD. Indeed, glutamate stimulates post-synaptic receptors involved in memory processes, while memantine decreases the excess of glutamate responsible for neuronal death in AD patients [39][40][41].
Other promising anti-AD agents are molecules preventing Aβ or tau aggregation, from small compounds [42][43][44] to peptides and monoclonal antibodies (mAbs) [14][44][45][46][47].
For example, based on the 17–21 residues of the Aβ peptide, i.e., LVFFA, Soto et al. designed a peptide inhibitor LPFFD, also known as iAβ5 [48][49]. Due to the substitution of valine with a proline residue, not able to fit in the β-sheet structure, iAβ5 exhibited a very low propensity to adopt a β-sheet conformation, preventing the interaction between Aβ molecules and the formation of β-sheet oligomers [48][50].
Moreover, residues 16–20 of the Aβ peptide, i.e., KLVFF, are crucial for the formation of β-sheet structures [51]. KLVFF peptide can bind its homologous sequence in Aβ, preventing its aggregation into amyloid fibrils [52][53][54].
An alternative strategy explored in AD treatment consists in the solubilization of preformed β-amyloid plaques, successfully realized with humanized mAbs or copper/zinc chelators. Indeed, oxidative damage – promoted by metals such as iron, zinc, copper, and aluminium – is one of the main causes of AD, since these metals interact with β-amyloids and promote their aggregation [55].
Therefore, the use of suitable chelators, such as the copper chelator D-penicillamine (Figure 1) [56][57], able to reduce the amount of metal in the brain, can represent a valid approach to AD treatment [58][59].
Further available anti-AD agents are non-steroidal anti-inflammatory drugs such as tarenflurbil (Figure 1), i.e., the pure R-enantiomer of flurbiprofen [60] and/or antioxidants [61], most of which are derived from natural sources.
1.2. Parkinson’s Disease
The second most common age-related neurodegenerative disorder and most common movement disorder is Parkinson’s disease (PD), with a prevalence of over 2% after the age of 65.
Pathological features of this ND are a reduced number of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies, i.e., eosinophilic intracellular inclusions formed by aggregates of the α-synuclein (SNCA) protein. These aggregates begin to form in the medulla and olfactory bulbs, spreading progressively—according to Braak’s six-stage description—to involve pons, midbrain, limbic lobe, amygdala, and neocortex [65][66][67][68]. The spread of Lewy bodies eventually leads to the dysfunction of other neurotransmitter systems, such as adrenergic, cholinergic, and serotonergic [69][70].
Owing to the geographical variability observed in PD incidence, this disease has been regarded for years as a purely sporadic disorder, mainly of environmental origin. The era of the recognized genetic contribution to the pathogenesis of PD began in the late 1990s, thanks to the Contursi family (from the namesake village in Campania, Italy), in which molecular genetic studies revealed in the gene locus 4q21-22 (termed PARK1) the first SNCA-associated PD [71]. Since this gene has been identified, more than twenty genes and several independent risk-associated variants have been linked to PD onset [72].
Several studies also demonstrated that the dysfunction of the UPS system plays a direct role in the pathogenesis of PD. Under physiological conditions, UPS is responsible for most of the protein turnover within cells. In PD, misfolded and aggregated SNCA impairs UPS function contributing to neuronal death [73].
It is now well accepted that PD is a complex multisystem ND, with both motor and non-motor signs and symptoms [74].
Since dopamine is involved in motor functions, its reduction results in brady-hypokinesia, rigidity, tremors, decreased balance, and gait difficulties, which are the cardinal features of the PD motor phenotype [75][76][77].
Non-motor signs—which are very prominent, especially in advanced PD stages—result from multiple neurotransmitter deficits in both the central and peripheral nervous systems, and significantly affect the quality of life of PD patients. These signs include: cognitive (dysexecutive syndrome, mild cognitive impairment to dementia), behavioural (hallucinations, delusions), mood depression, pain, dysautonomia (constipation, urgency, orthostatic hypotension), sleep, and vigilance disturbances [78][79]. Non-motor signs may also occur in the first stages of the disease, or precede the motor phase by several years, such as olfactory deficit due to olfactory nerve damage [80], or disturbances during rapid eye movement (REM) sleep, such as vivid dreams and/or nightmares, that may be responsible for self- or hetero-aggressive acts, or constipation.
Currently, the disease remains pathogenetically incurable, and only symptomatic approaches are available for PD patients, while no disease-modifying therapies have been described [81][82].
To restore the dopaminergic transmission, current approaches are essentially based on exogenous dopamine supply as such or administered as its levodopa (L-DOPA) precursor (Figure 2) [83][84].
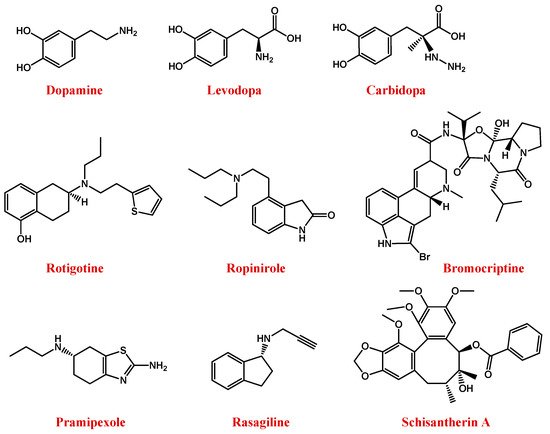
Figure 2. Molecular structures of main anti-PD drugs.
Indeed, dopamine is not able to overcome the blood–brain barrier (BBB), due to its low lipid solubility and lack of specific transporters. In contrast, its natural L-DOPA precursor can cross the BBB to a certain extent and is then converted to dopamine in the brain, due to the DOPA decarboxylase enzyme [85][86]. However, when orally administered, L-DOPA is rapidly decarboxylated to dopamine; thus, the amount of drug effectively able to reach as such the CNS is very small. So, to maintain its effectiveness, the L-DOPA dosage should be enhanced, but this increased dose is often associated with severe side effects, such as depression, anxiety, insomnia, agitation, nausea, and vomiting [70][81][87]. Moreover, its long-term use is accompanied by adverse effects, such as tardy action and disabling dyskinesia termed “Levodopa-induced dyskinesia” [88].
To maintain the L-DOPA efficiency avoiding high doses or high dosage frequency, another effective possibility consists in its co-administration with carbidopa (Figure 2), an inhibitor of the DOPA decarboxylase enzyme, able to prevent L-DOPA metabolism at the periphery [81][82].
However, disadvantages associated with the clinical use of L-DOPA stimulated the search for novel anti-PD drugs. In this context, rotigotine—a non-ergot-derived D3/D2/D1 agonist (Figure 2)—proved to have neuroprotective properties and lighten the motor symptoms of PD [89][90][91]. Alternative dopamine agonists are ropinirole, bromocriptine, and pramipexole (Figure 2) [91].
Ropinirole is a non-ergoline D2/D3 dopamine receptor agonist able to specifically bind D2-receptors in the striatum and substantia nigra [92][93].
Bromocriptine is a semi-synthetic ergopeptine derivative and a potent dopamine receptor agonist able to stimulate the striatal D2 non-adenyl cyclase-linked dopamine receptors [94].
Pramipexole is a non-ergot dopamine agonist especially used in association with L-DOPA or monoamine oxidase B (MAO-B) enzyme inhibitors [95][96].
Indeed, MAO-B is the main enzyme involved in the metabolic degradation of dopamine, reducing its available amount in the brain. Thus, the inhibition of its activity, using for example rasagiline (Figure 2), represents a useful approach to restore dopamine levels [97][98][99].
Urocortin, a corticotrophin-releasing hormone-related peptide, has recently been proposed as a cytoprotectant for cultured hippocampal neurons, cerebellar granule cells, and GABAergic neurons [100][101].
Iron accumulation in substantia nigra pars compacta has been proven to be a pathophysiological feature of PD, which could induce the death of dopaminergic neurons, reactive oxygen species (ROS) up-regulation, and further loss of motor control [102][103]. Thus, the term “iron-chelation therapy” generally refers to the reduction of abnormal iron accumulation in substantia nigra pars compacta [104].
1.3. Huntington’s Disease
Huntington’s disease (HD) is a rare neurodegenerative disorder that primarily affects basal ganglia neurons (caudate nucleus and putamen) with striatal medium spiny neurons (composed of GABAergic neurons) almost completely lost in the advanced stages of this disease. Early dysfunction with subsequent loss of cortical neurons is also prominent and consistent with a decrease in brain weight [107][108][109][110][111].
HD is caused by dominant mutations in the exon 1 of Huntingtin (HTT) gene encoding the HTT protein. The mutation consists of an abnormal repetition of the CAG nucleotide triplet (40 or above), leading to the production of an altered protein with an abnormally long polyglutamine tract (polyQ) at the N-terminal extremity [112]. This polyQ segment induces the intracellular aggregation of the mutant HTT protein (mHTT) in the caudate nucleus and putamen of basal ganglia, ultimately causing cortico-striatal dysfunction and degeneration [113][114][115][116][117].
The typical signs and symptoms of HD—which generally become evident between the ages of 30 and 50, with some reported cases of juvenile forms of HD (onset before the age of 20) [118]—consist of a triad of motor, cognitive and psychiatric symptoms. The most typical sign of HD is represented by choreic movements—from ancient Greek: χορεία (‘dance’)—hence, this ND is also known as Huntington’s chorea.
Other motor symptoms include abnormal postures (dystonia), rigidity with slow voluntary movements (bradykinesia), and convulsions. Cognitive impairments are featured by a progressive slowing of ideational processes and deterioration of memory, that gradually lead to dementia. Psychiatric symptoms, such as anxiety, apathy, loss of self-esteem, and guilt are very frequent in the first stages of the disease, and often precede motor symptoms. The suicide rate is higher in patients with early onset or clinically presymptomatic individuals. Other frequent and disabling signs of HD are weight loss, sleep disturbances, and loss of circadian rhythm [119].
According to macroscopic and microscopic examinations, pathological changes in HD were classified by Vonsattel into four grades (0–4), each correlated with the degree of clinical impairment determined at the last pre-death evaluation [120].
Multiple processes seem to relate CAG expansion to neurodegeneration in HD, including transcriptional factors and coactivators, ultimately leading to cell death [108][121]. In HD, oxidative stress and mitochondrial defects are also revealed, the latter ones also leading to neuronal loss and reduced activity of the electron transport chain. HD is also accompanied by neurochemical alterations in dopamine, adenosine, and glutamate receptors [121][122][123][124].
Antidopaminergic agents, e.g., tetrabenazine (TBZ, Figure 3), are the main drugs used for HD therapy [128][129]. Vesicular monoamine transporter 2 concentrates monoamines into vesicles, while TBZ reversibly inhibits this action, reducing presynaptic dopamine [128][130].
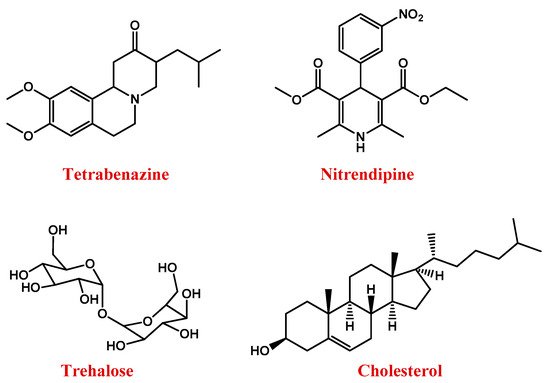
Figure 3. Molecular structures of main anti-HD drugs.
Nitrendipine (Figure 3) is a dihydropyridine calcium channel blocker that, exerting neuroprotective effects, can reduce the incidences of dementia in HD. However, being hydrophilic, it scarcely crosses the BBB [131].
Triggering a series of neuroprotective mechanisms, the disaccharide trehalose (Figure 3) can also improve cognitive performance in HD patients [132].
Several studies demonstrated that HD is also featured by abnormal brain cholesterol homeostasis [133][134]. Cholesterol dysregulation occurs in astrocytes [135][136] and is linked to the action of mHTT on sterol regulatory element-binding proteins (SREBPs) and its target genes, whose reduced transcription leads to lower production and a release of cholesterol in the brain. The low amount of cholesterol available to be taken up by neurons [137][138][139] impairs neuronal activities, causing brain malformations, and alterations in cognitive functions [140]. Therefore, strategies aimed at improving cholesterol delivery to neurons could be efficient in the treatment of the pathology [141][142].
On the other hand, neurotrophic factors are responsible for the growth, development, and survival of brain cells [143]. Changes in the levels and activities of neurotrophic factors, such as the brain-derived neurotrophic factor (BDNF), have been described in neurodegenerative disorders, including AD, PD, and HD [144][145][146][147]. Therefore, the use of genes that code BDNF can potentially prevent the death of brain cells and mitigate the symptoms of NDs [145][148].
Causes, symptoms, and conventional treatments of these three neurodegenerative diseases are schematically summarized in Figure 4.
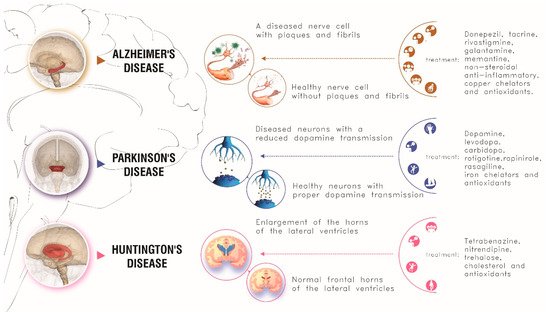
Figure 4. Main causes, symptoms and conventional treatments for the main three neurodegenerative disorders here described.
2. Oxidative Stress and Polyphenol Compounds in Neurodegenerative Diseases
For all the NDs above described, it has been reported that excessive oxidative stress is involved in neuronal damage [149]. Free radicals in the brain are formed as a consequence of oxidation processes or poor physiological antioxidant activity. In turn, ROS cause disruption in mitochondrial cellular lipids and proteins, as well as DNA damage, mitochondrial dysfunction, and genome instability [150][151][152][153].
Therefore, antioxidant agents have been widely investigated for their potential ability to prevent oxidative stress in NDs [154].
In this context, natural bioactive compounds, especially polyphenols, have gained increasing attention for their pharmacological and therapeutic potential [155][156][157][158][159].
A phenolic compound is a molecule having at least one aromatic ring on which one or more hydroxyl groups are attached. According to their main structural features, polyphenols can be classified as flavonoids and non-flavonoids with further subclasses [157][160][161].
Among polyphenols, the most important compounds are curcumin, resveratrol, rosmarinic acid, ferulic acid, α-mangostin, anthocyanins, epigallocatechin-3-gallate (EGCG), quercetin, and thymoquinone (Figure 5) [157].
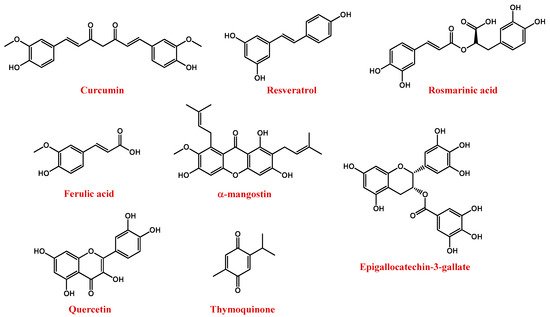
Figure 5. Molecular structures of polyphenol compounds here described.
Curcumin (Figure 5) is naturally present in turmeric (Curcuma longa), an important food and medication widely used in India and China. The chemical name of curcumin is diferuloylmethane and is a mixture of three major curcuminoids, i.e., curcumin, desmethoxy curcumin, and bis-desmethoxy curcumin [162].
Several studies proved that curcumin has antioxidant, anti-inflammatory, anticancer, antiviral, and antibacterial properties, thus exhibiting great potential in various diseases, including neurodegenerative ones [163][164][165]. Curcumin targets both β-amyloid and tau AD markers, decreasing the production of Aβ plaques, tau hyperphosphorylation and the formation of neurofibrillary tangles [166][167]. In the case of HD, curcumin showed beneficial effects, especially when added to the diet [168][169]. However, its poor bioavailability, resulting from its rapid metabolism and body clearance as well as poor BBB permeability, hindered its widespread use [170].
Resveratrol, or 3,5,4′-trihydroxystilbene (Figure 5), is a natural polyphenolic flavonoid and the main member of the stilbene family. It can be found in nature as both cis and trans isomers, the latter considered to be the most abundant and biologically active [171]. In particular, it is mainly present in the seeds and skins of grapes, red wine, mulberries, pomegranates, peanuts, tea, and rhubarb [172][173][174]. Resveratrol presents several effects, such as anti-cancer, anti-inflammatory, and anti-obesity [171]. The neuroprotective effects of resveratrol in NDs are related to its ability to reduce oxidative damage, toxicity, and apoptotic neuronal death [171][172][175].
Unfortunately, after intravenous injection, resveratrol is rapidly metabolized in the liver and intestine into both glucuronic acid and sulfate derivatives [176]. This results in a low resveratrol bioavailability which limits its pharmacological applications. In addition, resveratrol is also chemically unstable, since it is easily degraded by isomerization when exposed to elevated temperatures, pH changes, or UV light [171].
Rosmarinic acid (Figure 5) is a hydrophilic compound isolated from Rosmarinus officinalis L., which showed promising antioxidant properties linked to its ability to remove peroxynitrite anions and reduce inflammatory responses [177].
Ferulic acid (4-hydroxy-3-methoxycinnamic acid, Figure 5) exhibited both antioxidant and anti-inflammatory activities. In detail, it can reduce neuronal oxidative stress, preventing cell death [178][179].
α-mangostin (Figure 5) is a polyphenolic xanthone isolated from the pericarp, bark, and dried sap of Garcinia mangostana [180]. It exhibits a number of pharmacological effects, including neuroprotective, antioxidant, antitumour, and anti-neuroinflammatory actions [181][182][183][184].
Anthocyanins, polyphenolic compounds of the flavonoid family, have been found in fruits, grains, and flowers, and are reported to have antioxidant, anti-inflammatory, anti-apoptotic, and neuroprotective properties. Unfortunately, anthocyanins are chemically unstable, because their phenolic hydroxyl groups are easily oxidized to quinones with reduced biological activity [185][186][187].
EGCG (Figure 5), the major polyphenol in green tea, is known for its potent antioxidant properties [188]. In addition, it interacts with numerous proteins involved in NDs, such as Aβ amyloid, α-synuclein, and HTT [189][190][191].
Quercetin (Figure 5) is a dietary flavonoid with well-recognized antioxidant, anti-inflammatory and autophagy-inducer properties. Quercetin is present in apples, onions, parsley, berries, green tea, citrus fruits, and in some herbal remedies, e.g., ginkgo biloba [192]. For its potent neuroprotective action, quercetin represents a potential drug for the treatment of NDs [193][194][195].
Thymoquinone (2-isopropyl-5-methyl-1,2-benzoquinone, Figure 5) is the major active compound of the volatile oil of Nigella sativa seeds, featured by remarkable antioxidant, antitumour, anti-inflammatory, and immunomodulatory properties [196][197][198][199][200][201].
3. Targeting Brain:
Treatments of NDs are often clinically ineffective due to the poor accessibility of administered drugs to the desired site of action.
By engineering materials of a size usually within 1–100 nm, nanotechnology offers an alternative approach for promising and innovative therapeutic solutions in NDs. Nanoparticles can cross the BBB and release active molecules at target sites in the brain, minimizing side effects. This review focuses on the state-of-the-art of nanoengineered delivery systems for brain targeting in the treatment of AD, PD and HD.[202]
References
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035.
- Kanwar, J.R.; Sriramoju, B.; Kanwar, R.K. Neurological disorders and therapeutics targeted to surmount the blood-brain barrier. Int. J. Nanomed. 2012, 7, 3259–3278.
- Poovaiah, N.; Davoudi, Z.; Peng, H.; Schlichtmann, B.; Mallapragada, S.; Narasimhan, B.; Wang, Q. Treatment of neurodegenerative disorders through the blood-brain barrier using nanocarriers. Nanoscale 2018, 10, 16962–16983.
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 1–13.
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340.
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056.
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255.
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48.
- Allgaier, M.; Allgaier, C. An update on drug treatment options of Alzheimer’s disease. Front. Biosci. Landmark 2014, 19, 1345–1354.
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. J. R. Coll. Physicians Lond. 2016, 16, 247–253.
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, F1000.
- Husna Ibrahim, N.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 2020, 11, 261.
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72.
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508.
- Nisbet, R.M.; Götz, J. Amyloid-β and tau in Alzheimer’s disease: Novel pathomechanisms and non-pharmacological treatment strategies. J. Alzheimers Dis. 2018, 64, S517–S527.
- Gallardo, G.; Holtzman, D.M. Amyloid-β and tau at the crossroads of Alzheimer’s disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203.
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83.
- Ma, R.H.; Zhang, Y.; Hong, X.Y.; Zhang, J.F.; Wang, J.Z.; Liu, G.P. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 307–312.
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300.
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554.
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627.
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191.
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689.
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J. Alzheimers Dis. 2001, 3, 75–80.
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453.
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides—A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2011, 3, 303–312.
- Kumar, A.; Nisha, C.M.; Silakari, C.; Sharma, I.; Anusha, K.; Gupta, N.; Nair, P.; Tripathi, T.; Kumar, A. Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Formos. Med. Assoc. 2016, 115, 3–10.
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment combinations for Alzheimer’s disease: Current and future pharmacotherapy options. J. Alzheimers Dis. 2019, 67, 779–794.
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487.
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157.
- Christodoulou, C.; Melville, P.; Scherl, W.F.; MacAllister, W.S.; Elkins, L.E.; Krupp, L.B. Effects of donepezil on memory and cognition in multiple sclerosis. J. Neurol. Sci. 2006, 245, 127–136.
- Summers, W.K. Tacrine, and Alzheimer’s treatments. J. Alzheimers Dis. 2006, 9, 439–445.
- Birks, J.S.; Chong, L.Y.; Grimley Evans, J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, CD001191.
- Prvulovic, D.; Hampel, H.; Pantel, J. Galantamine for Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2010, 6, 345–354.
- Daulatzai, M.A. Pharmacotherpy and Alzheimer’s disease: The M-drugs (melatonin, minocycline, modafinil, and memantine) approach. Curr. Pharm. Des. 2016, 22, 2411–2430.
- Alam, S.; Lingenfelter, K.S.; Bender, A.M.; Lindsley, C.W. Classics in chemical neuroscience: Memantine. ACS Chem. Neurosci. 2017, 8, 1823–1829.
- Matsunaga, S.; Kishi, T.; Nomura, I.; Sakuma, K.; Okuya, M.; Ikuta, T.; Iwata, N. The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2018, 17, 1053–1061.
- Vicidomini, C.; Cioffi, F.; Broersen, K.; Roviello, V.; Riccardi, C.; Montesarchio, D.; Capasso, D.; Gaetano, S.D.; Roviello, G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019, 11, 285–302.
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62.
- Monteiro, K.L.C.; Alcântara, M.G.D.S.; de Aquino, T.M.; da Silva-Júnior, E.F. Tau protein aggregation in Alzheimer’s disease: Recent advances in the development of novel therapeutic agents. Curr. Pharm. Des. 2020, 26, 1682–1692.
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167.
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415.
- Pinheiro, L.; Faustino, C. Therapeutic strategies targeting amyloid-β in Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 418–452.
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680.
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826.
- Wood, S.J.; Wetzel, R.; Martin, J.D.; Hurle, M.R. Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide β/A4. Biochemistry 1995, 34, 724–730.
- Tjernberg, L.O.; Näslundt, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Tereniust, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548.
- Lowe, T.L.; Strzelec, A.; Kiessling, L.L.; Murphy, R.M. Structure-function relationships for inhibitors of β-Amyloid toxicity containing the recognition sequence KLVFF. Biochemistry 2001, 40, 7882–7889.
- Watanabe, K.I.; Nakamura, K.; Akikusa, S.; Okada, T.; Kodaka, M.; Okuno, H.; Watanabe, K.I.; Akikusa, S.; Konakahara, T. Inhibitors of fibril formation and cytotoxicity of β-amyloid peptide composed of KLVFF recognition element and flexible hydrophilic disrupting element. Biochem. Biophys. Res. Commun. 2002, 290, 121–124.
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. β-sheet breaker peptide prevents Aβ-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52.
- Lu, J.; Combs, G.F. Penicillamine: Pharmacokinetics and differential effects on zinc and copper status in chicks. J. Nutr. 1992, 122, 355–362.
- Cherny, R.A.; Barnham, K.J.; Lynch, T.; Volitakis, I.; Li, Q.X.; McLean, C.A.; Multhaup, G.; Beyreuther, K.; Tanzi, R.E.; Masters, C.L.; et al. Chelation and intercalation: Complementary properties in a compound for the treatment of Alzheimer’s disease. J. Struct. Biol. 2000, 130, 209–216.
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616.
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.S.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676.
- Gasparini, L.; Ongini, E.; Wenk, G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: Old and new mechanisms of action. J. Neurochem. 2004, 91, 521–536.
- Etminan, M.; Gill, S.; Samii, A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: Systematic review and meta-analysis of observational studies. Br. Med. J. 2003, 327, 128.
- Shah, S.A.; Lee, H.Y.; Bressan, R.A.; Yun, D.J.; Kim, M.O. Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death Dis. 2014, 5, e1026.
- Naseer, M.I.; Ullah, I.; Narasimhan, M.L.; Lee, H.Y.; Bressan, R.A.; Yoon, G.H.; Yun, D.J.; Kim, M.O. Neuroprotective effect of osmotin against ethanol-induced apoptotic neurodegeneration in the developing rat brain. Cell Death Dis. 2014, 5, e1150.
- Ali, T.; Yoon, G.H.; Shah, S.A.; Lee, H.Y.; Kim, M.O. Osmotin attenuates amyloid beta-induced memory impairment, tau phosphorylation and neurodegeneration in the mouse hippocampus. Sci. Rep. 2015, 5, 1–17.
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Radhakrishnan, D.M.; Goyal, V. Parkinson’s disease: A review. Neurol. India 2018, 66, S26–S35.
- Mehra, S.; Sahay, S.; Maji, S.K. α-synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 890–908.
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350.
- Rizzi, G.; Tan, K.R. Dopamine and acetylcholine, a circuit point of view in Parkinson’s disease. Front. Neural Circuits 2017, 11, 110.
- Hayes, M.W.; Fung, V.S.C.; Kimber, T.E.; O’Sullivan, J.D. Updates and advances in the treatment of Parkinson disease. Med. J. Aust. 2019, 211, 277–283.
- Polymeropoulos, M.H.; Lavedan, C.L.E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; Stenroos, E.S. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 12, 170–178.
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46.
- Archibald, N.; Miller, N.; Rochester, L. Neurorehabilitation in Parkinson disease. Handb. Clin. Neurol. 2013, 110, 435–442.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915.
- Alexoudi, A.; Alexoudi, I.; Gatzonis, S. Parkinson’s disease pathogenesis, evolution and alternative pathways: A review. Rev. Neurol. 2018, 174, 699–704.
- Meder, D.; Herz, D.M.; Rowe, J.B.; Lehéricy, S.; Siebner, H.R. The role of dopamine in the brain—lessons learned from Parkinson’s disease. Neuroimage 2019, 190, 79–93.
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474.
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov. Disord. 2015, 30, 1490–1504.
- Hawkes, C.H.; Shephard, B.C.; Daniel, S.E. Is Parkinson’s disease a primary olfactory disorder? QJM Mon. J. Assoc. Physicians 1999, 92, 473–480.
- Singh, N.; Pillay, V.; Choonara, Y.E. Advances in the treatment of Parkinson’s disease. Prog. Neurobiol. 2007, 81, 29–44.
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA J. Am. Med. Assoc. 2020, 323, 548–560.
- Whitfield, A.C.; Moore, B.T.; Daniels, R.N. Classics in chemical neuroscience: Levodopa. ACS Chem. Neurosci. 2014, 5, 1192–1197.
- Fahn, S.; Poewe, W. Levodopa: 50 years of a revolutionary drug for Parkinson disease. Mov. Disord. 2015, 30, 1–3.
- Nagatsua, T.; Sawadab, M. l-dopa therapy for Parkinson’s disease: Past, present, and future. Park. Relat. Disord. 2009, 15, S3–S8.
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and levodopa prodrugs for the treatment of Parkinson’s disease. Molecules 2018, 23, 40.
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s disease: Current status and future developments. Curr. Neuropharmacol. 2017, 16, 1239–1252.
- Lewitt, P.A. Levodopa therapy for Parkinson’s disease: Pharmacokinetics and pharmacodynamics. Mov. Disord. 2015, 30, 64–72.
- McAfee, D.A.; Hadgraft, J.; Lane, M.E. Rotigotine: The first new chemical entity for transdermal drug delivery. Eur. J. Pharm. Biopharm. 2014, 88, 586–593.
- Chen, F.; Jin, L.; Nie, Z. Safety and efficacy of rotigotine for treating Parkinson’s disease: A meta-analysis of randomised controlled trials. J. Pharm. Pharm. Sci. 2017, 20, 285–294.
- Contin, M.; Lopane, G.; Mohamed, S.; Calandra-Buonaura, G.; Capellari, S.; De Massis, P.; Nassetti, S.; Perrone, A.; Riva, R.; Sambati, L.; et al. Clinical pharmacokinetics of pramipexole, ropinirole and rotigotine in patients with Parkinson’s disease. Park. Relat. Disord. 2019, 61, 111–117.
- Pahwa, R.; Lyons, K.E.; Hauser, R.A. Ropinirole therapy for Parkinson’s disease. Expert Rev. Neurother. 2004, 4, 581–588.
- Jost, W.H.; Angersbach, D. Ropinirole, a non-ergoline dopamine agonist. CNS Drug Rev. 2005, 11, 253–272.
- Lieberman, A.N.; Goldstein, M. Bromocriptine in Parkinson disease. Pharmacol. Rev. 1985, 37, 217–227.
- Antonini, A.; Barone, P.; Ceravolo, R.; Fabbrini, G.; Tinazzi, M.; Abbruzzese, G. Role of pramipexole in the management of Parkinsons disease. CNS Drugs 2010, 24, 829–841.
- Shen, T.; Ye, R.; Zhang, B. Efficacy and safety of pramipexole extended-release in Parkinson’s disease: A review based on meta-analysis of randomized controlled trials. Eur. J. Neurol. 2017, 24, 835–843.
- Chen, J.J.; Swope, D.M.; Dashtipour, K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007, 29, 1825–1849.
- McCormack, P.L. Rasagiline: A review of its use in the treatment of idiopathic parkinson’s disease. CNS Drugs 2014, 28, 1083–1097.
- Schettino, C.; Dato, C.; Capaldo, G.; Sampaolo, S.; Di Iorio, G.; Melone, M.A.B. Rasagiline for sleep disorders in patients with Parkinson’s disease: A prospective observational study. Neuropsychiatr. Dis. Treat. 2016, 12, 2497–2502.
- Facci, L.; Stevens, D.A.; Pangallo, M.; Franceschini, D.; Skaper, S.D.; Strijbos, P.J.L.M. Corticotropin-releasing factor (CRF) and related peptides confer neuroprotection via type 1 CRF receptors. Neuropharmacology 2003, 45, 623–636.
- Choi, J.S.; Pham, T.T.H.; Jang, Y.J.; Bao, C.B.; Lee, B.H.; Joo, K.M.; Cha, C.I.; Lee, K.H. Corticotropin-releasing factor (CRF) and urocortin promote the survival of cultured cerebellar GABAergic neurons through the Type 1 CRF receptor. J. Korean Med. Sci. 2006, 21, 518–526.
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov. Disord. 2010, 25, S63–S70.
- Das, B.; Vedachalam, S.; Luo, D.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Development of a highly pPotent D2/D3 agonist and a partial agonist from structure-activity relationship study of N6-(2-(4-(1H-indol-5-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzothiazole-2,6-diamine analogues: Implication in the treatmen. J. Med. Chem. 2015, 58, 9179–9195.
- Xu, Q.; Kanthasamy, A.G.; Reddy, M.B. Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson’s disease. Toxicology 2008, 245, 101–108.
- Zhang, L.Q.; Sa, F.; Chong, C.M.; Wang, Y.; Zhou, Z.Y.; Chang, R.C.C.; Chan, S.W.; Hoi, P.M.; Yuen Lee, S.M. Schisantherin A protects against 6-OHDA-induced dopaminergic neuron damage in zebrafish and cytotoxicity in SH-SY5Y cells through the ROS/NO and AKT/GSK3β pathways. J. Ethnopharmacol. 2015, 170, 8–15.
- Sa, F.; Zhang, L.Q.; Chong, C.M.; Guo, B.J.; Li, S.; Zhang, Z.J.; Zheng, Y.; Hoi, P.M.; Lee, S.M.Y. Discovery of novel anti-parkinsonian effect of schisantherin A in In Vitro and In Vivo. Neurosci. Lett. 2015, 593, 7–12.
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930.
- Melone, M.A.B.; Jori, F.; Peluso, G. Huntingtons disease: New frontiers for molecular and cell therapy. Curr. Drug Targets 2005, 6, 43–56.
- Zuccato, C.; Cattaneo, E. Huntington’s disease. Handb. Exp. Pharmacol. 2015, 220, 357–409.
- Zuccato, C.; Cattaneo, E. The Huntington’s paradox. Sci. Am. 2016, 315, 56–61.
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138.
- Cattaneo, E.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Squitieri, F.; Sipione, S. Loss of normal huntingtin function: New developments in Huntington’s disease research. Trends Neurosci. 2001, 24, 182–188.
- Cattaneo, E. Dysfunction of wild-type huntingtin in Huntington disease. News Physiol. Sci. 2003, 18, 34–37.
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981.
- Melone, M.A.B.; Calarco, A.; Petillo, O.; Margarucci, S.; Colucci-D’Amato, L.; Galderisi, U.; Koverech, G.; Peluso, G. Mutant huntingtin regulates EGF receptor fate in non-neuronal cells lacking wild-type protein. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 105–113.
- Kay, C.; Collins, J.A.; Wright, G.E.B.; Baine, F.; Miedzybrodzka, Z.; Aminkeng, F.; Semaka, A.J.; McDonald, C.; Davidson, M.; Madore, S.J.; et al. The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 346–357.
- Nuzzo, M.T.; Fiocchetti, M.; Totta, P.; Melone, M.A.B.; Cardinale, A.; Fusco, F.R.; Gustincich, S.; Persichetti, F.; Ascenzi, P.; Marino, M. Huntingtin polyQ mutation impairs the 17β-estradiol/neuroglobin pathway devoted to neuron survival. Mol. Neurobiol. 2017, 54, 6634–6646.
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005.
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A mitochondria-associated oxidative stress perspective on Huntington’s disease. Front. Mol. Neurosci. 2018, 11, 329.
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P. Neuropathological classification of huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577.
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98.
- Anzilotti, S.; Giampà, C.; Laurenti, D.; Perrone, L.; Bernardi, G.; Melone, M.A.B.; Fusco, F.R. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res. Bull. 2012, 87, 350–358.
- Leuti, A.; Laurenti, D.; Giampà, C.; Montagna, E.; Dato, C.; Anzilotti, S.; Melone, M.A.B.; Bernardi, G.; Fusco, F.R. Phosphodiesterase 10A (PDE10A) localization in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2013, 52, 104–116.
- Cardinale, A.; Fusco, F.R.; Paldino, E.; Giampà, C.; Marino, M.; Nuzzo, M.T.; D’Angelo, V.; Laurenti, D.; Straccia, G.; Fasano, D.; et al. Localization of neuroglobin in the brain of R6/2 mouse model of Huntington’s disease. Neurol. Sci. 2018, 39, 275–285.
- Perrone, L.; Melone, M.A.B. New targets for therapy in polyglutamine (polyQ) expansion diseases. Curr. Drug Ther. 2008, 3, 177–189.
- Fusco, F.R.; Anzilotti, S.; Giampà, C.; Dato, C.; Laurenti, D.; Leuti, A.; Colucci D’Amato, L.; Perrone, L.; Bernardi, G.; Melone, M.A.B. Changes in the expression of extracellular regulated kinase (ERK 1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol. Dis. 2012, 46, 225–233.
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 5, 1–8.
- Venuto, C.S.; Mcgarry, A.; Ma, Q.; Kieburtz, K. Pharmacologic approaches to the treatment of Huntington’s disease. Mov. Disord. 2012, 27, 31–41.
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750.
- Poon, L.H.; Kang, G.A.; Lee, A.J. Role of tetrabenazine for Huntington’s disease-associated chorea. Ann. Pharmacother. 2010, 44, 1080–1089.
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305.
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154.
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.D.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhem, I.; et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131, 2851–2859.
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S. Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol. Dis. 2013, 55, 37–43.
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015, 22, 690–702.
- Valenza, M.; Leoni, V.; Karasinska, J.M.; Petricca, L.; Fan, J.; Carroll, J.; Pouladi, M.A.; Fossale, E.; Nguyen, H.P.; Riess, O.; et al. Cholesterol defect is marked across multiple rodent models of Huntington’s disease and is manifest in astrocytes. J. Neurosci. 2010, 30, 10844–10850.
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. 2005, 25, 9932–9939.
- Valenza, M.; Leoni, V.; Tarditi, A.; Mariotti, C.; Björkhem, I.; Di Donato, S.; Cattaneo, E. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2007, 28, 133–142.
- Valenza, M.; Carroll, J.B.; Leoni, V.; Bertram, L.N.; Björkhem, I.; Singaraja, R.R.; Di Donato, S.; Lutjohann, D.; Hayden, M.R.; Cattaneo, E. Cholesterol biosynthesis pathway is disturbed in YAC128 mice and is modulated by huntingtin mutation. Hum. Mol. Genet. 2007, 16, 2187–2198.
- Valenza, M.; Cattaneo, E. Cholesterol dysfunction in neurodegenerative diseases: Is Huntington’s disease in the list? Prog. Neurobiol. 2006, 80, 165–176.
- Valenza, M.; Cattaneo, E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011, 6, 919–930.
- Valenza, M.; Cattaneo, E. Neuroprotection and brain cholesterol biosynthesis in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, E143.
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123–131.
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330.
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322.
- Giampà, C.; Montagna, E.; Dato, C.; Melone, M.A.B.; Bernardi, G.; Fusco, F.R. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2013, 8, e64037.
- Axelsen, T.M.; Woldbye, D.P.D. Gene therapy for Parkinson’s disease, an update. J. Parkinsons. Dis. 2018, 8, 195–215.
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175.
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967.
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214.
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125.
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583.
- Linseman, D.A. Therapeutic antioxidants for neurodegenerative disease. Recent Pat. CNS Drug Discov. 2012, 7, 183.
- Vidoni, C.; Castiglioni, A.; Seca, C.; Secomandi, E.; Melone, M.A.B.; Isidoro, C. Dopamine exacerbates mutant Huntingtin toxicity via oxidative-mediated inhibition of autophagy in SH-SY5Y neuroblastoma cells: Beneficial effects of anti-oxidant therapeutics. Neurochem. Int. 2016, 101, 132–143.
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317.
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderisi, U.; Di Iorio, G.; Giordano, A.; Melone, M.A.B. Adult-onset brain tumors and neurodegeneration: Are polyphenols protective? J. Cell. Physiol. 2018, 233, 3955–3967.
- Perrone, L.; Sampaolo, S.; Melone, M.A.B. Bioactive phenolic compounds in the modulation of central and peripheral nervous system cancers: Facts and misdeeds. Cancers 2020, 12, 454.
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ahmad, J.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M.; Aleya, L. Neuroprotective role of polyphenols against oxidative stress-mediated neurodegeneration. Eur. J. Pharmacol. 2020, 886, 173412.
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892.
- Fraga, C.G.; Croft, K.D.; Kennedy, D.O.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528.
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773.
- Perrone, D.; Ardito, F.; Giannatempo, G.; Dioguardi, M.; Troiano, G.; Lo Russo, L.; De Lillo, A.; Laino, L.; Lo Muzio, L. Biological and therapeutic activities, and anticancer properties of curcumin. Exp. Ther. Med. 2015, 10, 1615–1623.
- Stanić, Z. Curcumin, a compound from natural sources, a true scientific challenge—A review. Plant Foods Hum. Nutr. 2017, 72, 1–12.
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The autophagy signaling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients 2019, 11, 1881.
- Goozee, K.G.; Shah, T.M.; Sohrabi, H.R.; Rainey-Smith, S.R.; Brown, B.; Verdile, G.; Martins, R.N. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr. 2016, 115, 449–465.
- Tang, M.; Taghibiglou, C.; Liu, J. The mechanisms of action of curcumin in Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 1003–1016.
- Hickey, M.A.; Zhu, C.; Medvedeva, V.; Lerner, R.P.; Patassini, S.; Franich, N.R.; Maiti, P.; Frautschy, S.A.; Zeitlin, S.; Levine, M.S.; et al. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 2012, 7, 1–16.
- Finicelli, M.; Squillaro, T.; Di Cristo, F.; Di Salle, A.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Metabolic syndrome, mediterranean diet, and polyphenols: Evidence and perspectives. J. Cell. Physiol. 2019, 234, 5807–5826.
- Yang, K.Y.; Lin, L.C.; Tseng, T.Y.; Wang, S.C.; Tsai, T.H. Oral bioavailability of curcumin in rat and the herbal analysis from curcuma longa by LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 853, 183–189.
- R Neves, A.; Lucio, M.; LC Lima, J.; Reis, S. Resveratrol in medicinal chemistry: A critical review of its pharmacokinetics, drug-delivery, and membrane interactions. Curr. Med. Chem. 2012, 19, 1663–1681.
- Markus, M.A.; Morris, B.J. Resveratrol in prevention and treatment of common clinical conditions of aging. Clin. Interv. Aging 2008, 3, 331–339.
- Wu, C.F.; Yang, J.Y.; Wang, F.; Wang, X.X. Resveratrol: Botanical origin, pharmacological activity and applications. Chin. J. Nat. Med. 2013, 11, 1–15.
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A focus on several neurodegenerative diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169.
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.A.B.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187.
- Anekonda, T.S. Resveratrol-A boon for treating Alzheimer’s disease? Brain Res. Rev. 2006, 52, 316–326.
- Alkam, T.; Nitta, A.; Mizoguchi, H.; Itoh, A.; Nabeshima, T. A natural scavenger of peroxynitrites, rosmarinic acid, protects against impairment of memory induced by Aβ(25–35). Behav. Brain Res. 2007, 180, 139–145.
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic acid: Therapeutic potential through its antioxidant property. J. Clin. Biochem. Nutr. 2007, 40, 92–100.
- Mancuso, C.; Santangelo, R. Ferulic acid: Pharmacological and toxicological aspects. Food Chem. Toxicol. 2014, 65, 185–195.
- Chen, G.; Li, Y.; Wang, W.; Deng, L. Bioactivity and pharmacological properties of α-mangostin from the mangosteen fruit: A review. Expert Opin. Ther. Pat. 2018, 28, 415–427.
- Chen, L.G.; Yang, L.L.; Wang, C.C. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem. Toxicol. 2008, 46, 688–693.
- Pedraza-Chaverrí, J.; Reyes-Fermín, L.M.; Nolasco-Amaya, E.G.; Orozco-Ibarra, M.; Medina-Campos, O.N.; González-Cuahutencos, O.; Rivero-Cruz, I.; Mata, R. ROS scavenging capacity and neuroprotective effect of α-mangostin against 3-nitropropionic acid in cerebellar granule neurons. Exp. Toxicol. Pathol. 2009, 61, 491–501.
- Wang, Y.; Xia, Z.; Xu, J.R.; Wang, Y.X.; Hou, L.N.; Qiu, Y.; Chen, H.Z. α-mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates β-amyloid oligomers-induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology 2012, 62, 871–881.
- Do, H.T.T.; Cho, J. Mangosteen pericarp and its bioactive xanthones: Potential therapeutic value in Alzheimer’s disease, Parkinson’s disease, and depression with pharmacokinetic and safety profiles. Int. J. Mol. Sci. 2020, 21, 6211.
- Giusti, M.M.; Wrolstad, R.E. Acylated anthocyanins from edible sources and their applications in food systems. Biochem. Eng. J. 2003, 14, 217–225.
- Zafra-Stone, S.; Yasmin, T.; Bagchi, M.; Chatterjee, A.; Vinson, J.A.; Bagchi, D. Berry anthocyanins as novel antioxidants in human health and disease prevention. Mol. Nutr. Food Res. 2007, 51, 675–683.
- Ghosh, D.; Konishi, T. Anthocyanins and anthocyanin-rich extracts: Role in diabetes and eye function. Asia Pac. J. Clin. Nutr. 2007, 16, 200–208.
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B.H. Neuroprotective molecular mechanisms of (-)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr. 2009, 4, 283–296.
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751.
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187.
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715.
- D’Andrea, G. Quercetin: A flavonol with multifaceted therapeutic applications? Fitoterapia 2015, 106, 256–271.
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of neuroprotection by quercetin: Counteracting oxidative stress and more. Oxidative Med. Cell. Longev. 2016, 2016, 2986796.
- Elumalai, P.; Lakshmi, S. Role of quercetin benefits in neurodegeneration. Adv. Neurobiol. 2016, 12, 229–245.
- Amanzadeh, E.; Esmaeili, A.; Rahgozar, S.; Nourbakhshnia, M. Application of quercetin in neurological disorders: From nutrition to nanomedicine. Rev. Neurosci. 2019, 30, 555–572.
- Kanter, M. Nigella sativa and derived thymoquinone prevents hippocampal neurodegeneration after chronic toluene exposure in rats. Neurochem. Res. 2008, 33, 579–588.
- Radad, K.; Moldzio, R.; Taha, M.; Rausch, W.D. Thymoquinone protects dopaminergic neurons against MPP+ and rotenone. Phyther. Res. 2009, 23, 696–700.
- Abdelmeguid, N.E.; Fakhoury, R.; Kamal, S.M.; Al Wafai, R.J. Effects of Nigella sativa and thymoquinone on biochemical and subcellular changes in pancreatic β-cells of streptozotocin-induced diabetic rats. J. Diabetes 2010, 84, 127–134.
- Gilhotra, N.; Dhingra, D. Thymoquinone produced antianxiety-like effects in mice through modulation of GABA and NO levels. Pharmacol. Rep. 2011, 63, 660–669.
- Islam, F.; Khan, A.; Vaibhav, K.; Javed, H.; Moshahid Khan, M.; Tabassum, R.; Ahmed, M.E.; Srivastava, P.; Khuwaja, G.; Islam, F.; et al. Attenuation of Aβ-induced neurotoxicity by thymoquinone via inhibition of mitochondrial dysfunction and oxidative stress. Mol. Cell. Biochem. 2012, 369, 55–65.
- Ismail, N.; Ismail, M.; Azmi, N.H.; Abu Bakar, M.F.; Basri, H.; Abdullah, M.A. Modulation of hydrogen peroxide-induced oxidative stress in human neuronal cells by thymoquinone-rich fraction and thymoquinone via transcriptomic regulation of antioxidant and apoptotic signaling genes. Oxidative Med. Cell. Longev. 2016, 2016, 2528935.
- Claudia Riccardi; Filomena Napolitano; Daniela Montesarchio; Simone Sampaolo; Mariarosa Anna Beatrice Melone; Nanoparticle-Guided Brain Drug Delivery: Expanding the Therapeutic Approach to Neurodegenerative Diseases. Pharmaceutics 2021, 13, 1897, 10.3390/pharmaceutics13111897.
More