Many researchers have extensively explored selective hydrogenation of 5-hydroxymethylfurfural (HMF to DMF) to 2,5-dimethylfuran (DMF), and some of them were able to achieve high conversion and a high yield of DMF under certain reaction conditions. However, the main challenge is to reduce the operational cost by using cost-effective catalysts as an alternative to the expensive noble metals. Overall, the approach of using bifunctional catalysts is an important direction for catalysis in general and particularly for reactions that require significantly different types of elementary steps as part of an overall reaction. For this reason, efforts to use the rational design of interfaces will become important for supplementing the promising initial work in demonstrating bifunctional catalysis for catalytic transformations of biomass-derived 5-hydroxymethylfurfural to a potential liquid fuel, 2,5-dimethylfuran. Apart from the different types of metal, it was also observed that reaction conditions, namely the type of solvent, H2 donor, reaction temperature, reaction time and H2.
- hydroxymethylfurfural
- dimethylfuran
- hydrodeoxygenation
- Catalysts
1. Introduction
2. Hydrogenation with Noble Metal Catalysts
3. Hydrogenation Reactions with a Transitional Metal Catalyst
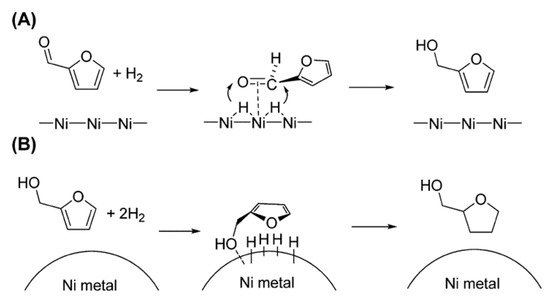
4. Hydrogenation Reactions with Bimetallic Catalysts
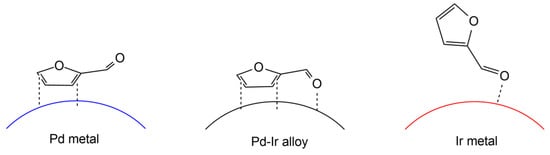
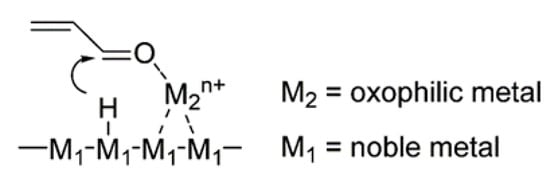
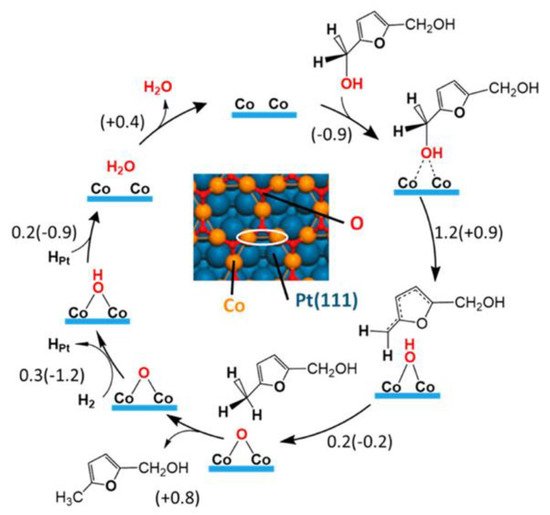
5. The Role of a Support
6. The Role of Solvents
7. Conclusions
References
- De, S.; Dutta, S.; Saha, B. One-Pot Conversions of Lignocellulosic and Algal Biomass into Liquid Fuels. ChemSusChem 2012, 5, 1826–1833.
- Hu, L.; Tang, X.; Xu, J.; Wu, Z.; Lin, L.; Liu, S. Selective Transformation of 5-Hydroxymethylfurfural into the Liquid Fuel 2,5-Dimethylfuran over Carbon-Supported Ruthenium. Ind. Eng. Chem. Res. 2014, 53, 3056–3064.
- Mitra, J.; Zhou, X.; Rauchfuss, T. Pd/C-catalyzed reactions of HMF: Decarbonylation, hydrogenation, and hydrogenolysis. Green Chem. 2014, 17, 307–313.
- Zhang, J.; Lin, L.; Liu, S. Efficient Production of Furan Derivatives from a Sugar Mixture by Catalytic Process. Energy Fuels 2012, 26, 4560–4567.
- Shun, N.; Naoya, I.; Kohki, E. Selective hydrogenation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-dimethylfuran (DMF) under atmospheric hydrogen pressure over carbon supported PdAu bimetallic catalyst. Catalysis Today 2014, 232, 89–98.
- Thananatthanachon, T.; Rauchfuss, T.B. Efficient Production of the Liquid Fuel 2,5-Dimethylfuran from Fructose Using Formic Acid as a Reagent. Angew. Chem. Int. Ed. 2010, 49, 6616–6618.
- Dutta, S.; Mascal, M. Novel Pathways to 2,5-Dimethylfuran via Biomass-Derived 5-(Chloromethyl)furfural. ChemSusChem 2014, 7, 3028–3030.
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558.
- Chatterjee, M.; Ishizaka, T.; Kawanami, H. Hydrogenation of 5-hydroxymethylfurfural in supercritical carbon diox-ide-water: A tunable approach to dimethylfuran selectivity. Green Chem. 2014, 16, 1543–1551.
- Lucas, N.; Kanna, N.R.; Nagpure, A.S.; Kokate, G.; Chilukuri, S. Novel catalysts for valorization of biomass to value-added chemicals and fuels. J. Chem. Sci. 2014, 126, 403–413.
- Zu, Y.; Yang, P.; Wang, J.; Liu, X.; Ren, J.; Lu, G.; Wang, Y. Efficient production of the liquid fuel 2,5-dimethylfuran from 5-hydroxymethylfurfural over Ru/Co3O4 catalyst. Appl. Catal. B Environ. 2014, 146, 244–248.
- Wang, G.-H.; Hilgert, J.; Richter, F.H.; Wang, F.R.; Bongard, H.; Spliethoff, B.; Weidenthaler, C.; Schüth, F. Platinum–cobalt bimetallic nanoparticles in hollow carbon nanospheres for hydrogenolysis of 5-hydroxymethylfurfural. Nat. Mater. 2014, 13, 293–300.
- Saha, B.; Abu-Omar, M.M. Current Technologies, Economics, and Perspectives for 2,5-Dimethylfuran Production from Biomass-Derived Intermediates. ChemSusChem 2015, 8, 1133–1142.
- Kong, X.; Zheng, R.; Zhu, Y.; Ding, G.; Zhu, Y.; Li, Y.-W. Rational design of Ni-based catalysts derived from hydrotalcite for selective hydrogenation of 5-hydroxymethylfurfural. Green Chem. 2015, 17, 2504–2514.
- Nakagawa, Y.; Tamura, M.; Tomishige, K. Catalytic Reduction of Biomass-Derived Furanic Compounds with Hydrogen. ACS Catal. 2013, 3, 2655–2668.
- Fouilloux, P. The nature of raney nickel, its adsorbed hydrogen and its catalytic activity for hydrogenation reactions (review). Appl. Catal. 1983, 8, 1–42.
- Iriondo, A.; Mendiguren, A.; Güemez, M.; Requies, J.; Cambra, J.F. 2,5-DMF production through hydrogenation of real and synthetic 5-HMF over transition metal catalysts supported on carriers with different nature. Catal. Today 2017, 279, 286–295.
- Nagpure, A.S.; Venugopal, A.K.; Lucas, N.; Manikandan, M.; Thirumalaiswamy, R.; Chilukuri, S. Renewable fuels from biomass-derived compounds: Ru-containing hydrotalcites as catalysts for conver-sion of HMF to 2,5-dimethylfuran. Catal. Sci. Technol. 2015, 5, 1463–1472.
- Vetere, V.; Merlo, A.B.; Ruggera, J.F.; Casella, M.L. Transition metal-based bimetallic catalysts for the chemoselective hydrogenation of furfuraldehyde. J. Braz. Chem. Soc. 2010, 21, 914–920.
- Nakagawa, Y.; Nakazawa, H.; Watanabe, H.; Tomishige, K. Total Hydrogenation of Furfural over a Silica-Supported Nickel Catalyst Prepared by the Reduction of a Nickel Nitrate Precursor. ChemCatChem 2012, 4, 1791–1797.
- Sugano, Y.; Shiraishi, Y.; Tsukamoto, D.; Ichikawa, S.; Tanaka, S.; Hirai, T. Supported Au–Cu Bimetallic Alloy Nanoparticles: An Aerobic Oxidation Catalyst with Regenerable Activity by Visible-Light Irradiation. Angew. Chem. Int. Ed. 2013, 52, 5295–5299.
- Iglesia, E.; Soled, S.; Fiato, R.; Via, G. Bimetallic Synergy in Cobalt Ruthenium Fischer-Tropsch Synthesis Catalysts. J. Catal. 1993, 143, 345–368.
- Zhu, L.; Zheng, L.; Du, K.; Fu, H.; Li, Y.; You, G.; Chen, B.H. An efficient and stable Ru-Ni/C nano-bimetallic catalyst with a comparatively low Ru loading for benzene hydrogenation under mild reaction conditions. RSC Adv. 2013, 3, 713–719.
- Ruban, A.V.; Skriver, H.L.; Nørskov, J.K. Surface segregation energies in transition-metal alloys. Phys. Rev. B 1999, 59, 15990–16000.
- Yoon, B.; Pan, H.-B.; Wai, C.M. Relative Catalytic Activities of Carbon Nanotube-Supported Metallic Nanoparticles for Room-Temperature Hydrogenation of Benzene. J. Phys. Chem. C 2009, 113, 1520–1525.
- Oosthuizen, R.S.; Nyamori, V.O. Carbon Nanotubes as Supports for Palladium and Bimetallic Catalysts for Use in Hydrogenation Reactions. Platin. Met. Rev. 2011, 55, 154–169.
- Li, H.; Li, H.; Deng, J.-F. Glucose hydrogenation over Ni–B/SiO2 amorphous alloy catalyst and the promoting effect of metal dopants. Catal. Today 2002, 74, 53–63.
- Jordão, M.H.; Simões, V.; Cardoso, D. Zeolite supported Pt-Ni catalysts in n-hexane isomerization. Appl. Catal. A: Gen. 2007, 319, 1–6.
- Scholz, D.; Aellig, C.; Hermans, I. Catalytic Transfer Hydrogenation/Hydrogenolysis for Reductive Upgrading of Furfural and 5-(Hydroxymethyl)furfural. ChemSusChem 2013, 7, 268–275.
- Chen, B.; Li, F.; Huang, Z.; Yuan, G. Carbon-coated Cu-Co bimetallic nanoparticles as selective and recyclable catalysts for production of biofuel 2,5-dimethylfuran. Appl. Catal. B Environ. 2017, 200, 192–199.
- Luo, J.; Lee, J.D.; Yun, H.; Wang, C.; Monai, M.; Murray, C.B.; Fornasiero, P.; Gorte, R.J. Base metal-Pt alloys: A general route to high selectivity and stability in the production of biofuels from HMF. Appl. Catal. B Environ. 2016, 199, 439–446.
- Yu, L.; He, L.; Chen, J.; Zheng, J.; Ye, L.; Lin, H.; Yuan, Y. Robust and Recyclable Nonprecious Bimetallic Nanoparticles on Carbon Nanotubes for the Hydrogenation and Hydrogenolysis of 5-Hydroxymethylfurfural. ChemCatChem 2015, 7, 1701–1707.
- Bottari, G.; Kumalaputri, A.J.; Krawczyk, K.; Feringa, B.L.; Heeres, H.J.; Barta, K. Copper-Zinc Alloy Nanopowder: A Robust Precious-Metal-Free Catalyst for the Conversion of 5-Hydroxymethylfurfural. ChemSusChem 2015, 8, 1323–1327.
- Sun, J.; Yang, G.; Ma, Q.; Ooki, I.; Taguchi, A.; Abe, T.; Xie, Q.; Yoneyama, Y.; Tsubaki, N. Fabrication of active Cu–Zn nanoalloys on H-ZSM5 zeolite for enhanced dimethyl ether synthesis via syngas. J. Mater. Chem. A 2014, 2, 8637–8643.
- Nakagawa, Y.; Takada, K.; Tamura, M.; Tomishige, K. Total Hydrogenation of Furfural and 5-Hydroxymethylfurfural over Supported Pd–Ir Alloy Catalyst. ACS Catal. 2014, 4, 2718–2726.
- Ni, Z.-M.; Xia, M.Y.; Shi, W.; Qian, P.P. Adsorption and Decarbonylation Reaction of Furfural on Pt (111) Surface. Acta Phys. -Chim. Sinica 2013, 29, 1916–1922.
- Aramendía, M.; Borau, V.; Jiménez, C.; Marinas, J.; Porras, A.; Urbano, F.J. Selective Liquid-Phase Hydrogenation of Citral over Supported Palladium. J. Catal. 1997, 172, 46–54.
- Vicente, A.; Lafaye, G.; Especel, C.; Marécot, P.; Williams, C.T. The relationship between the structural properties of bimetallic Pd–Sn/SiO 2 catalysts and their performance for selective citral hydrogenation. J. Catal. 2011, 283, 133–142.
- Luo, J.; Yun, H.; Mironenko, A.V.; Goulas, K.; Lee, J.D.; Monai, M.; Wang, C.; Vorotnikov, V.; Murray, C.B.; Vlachos, D.G.; et al. Mechanisms for High Selectivity in the Hydrodeoxygenation of 5-Hydroxymethylfurfural over PtCo Nanocrystals. ACS Catalysis 2016, 6, 4095–4104.
- Serp, P.; Figueiredo, J.L. (Eds.) Carbon Materials for Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2009.
- Priecel, P.; Endot, N.A.; Carà, P.D.; Lopez-Sanchez, J.A. Fast Catalytic Hydrogenation of 2,5-Hydroxymethylfurfural to 2,5-Dimethylfuran with Ruthenium on Car-bon Nanotubes. Ind. Eng. Chem. Res. 2018, 57, 1991–2002.
- Alamillo, R.; Tucker, M.; Chia, M.; Pagan-Torres, Y.; Dumesic, J. The selective hydrogenation of biomass-derived 5-hydroxymethylfurfural using heterogeneous catalysts. Green Chem. 2012, 14, 1413–1419.
- Cai, H.; Li, C.; Wang, A.; Zhang, T. Biomass into chemicals: One-pot production of furan-based diols from carbohydrates via tandem reactions. Catal. Today 2014, 234, 59–65.
- Ohyama, J.; Esaki, A.; Yamamoto, Y.; Arai, S.; Satsuma, A. Selective hydrogenation of 2-hydroxymethyl-5-furfural to 2,5-bis(hydroxymethyl)furan over gold sub-nano clusters. RSC Adv. 2013, 3, 1033–1036.
- Mäki-Arvela, P.; Ruiz, D.; Murzin, D.Y. Catalytic Hydrogenation/Hydrogenolysis of 5-Hydroxymethylfurfural to 2,5-Dimethylfuran. ChemSusChem. 2020, 14, 150–168.
- Esteves, L.M.; Brijaldo, M.H.; Oliveira, E.G.; Martínez, J.J.; Rojas, H.; Caytuero, A.; Passos, F.B. Effect of support on selective 5-hydroxymethylfurfural hydrogenation towards 2,5-dimethylfuran over copper catalysts. Fuel 2020, 270, 117524.
- Jae, J.; Zheng, W.; Karim, A.M.; Wei, G.; Lobo, R.F.; Vlachos, D.G. The Role of Ru and RuO2in the Catalytic Transfer Hydrogenation of 5-Hydroxymethylfurfural for the Production of 2,5-Dimethylfuran. ChemCatChem 2014, 6, 848–856.
- Chatterjee, M.; Ishizaka, T.; Kawanami, H. Selective hydrogenation of 5-hydroxymethylfurfural to 2,5-bis-(hydroxymethyl)furan using Pt/MCM-41 in an aqueous medium: A simple approach. Green Chem. 2014, 16, 4734–4739.
- Wang, H.; Zhu, C.; Li, D.; Liu, Q.; Tan, J.; Wang, C.; Cai, C.; Ma, L. Recent advances in catalytic conversion of biomass to 5-hydroxymethylfurfural and 2, 5-dimethylfuran. Renew. Sustain. Energy Rev. 2019, 103, 227–247.