Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Vivi Li and Version 2 by Alok Paul.
Opioids are broadly used as therapeutic agents against moderate to severe acute and chronic pain. Noticeably, these analgesics have many limitations as they induce analgesic tolerance, addiction, physical dependence, respiratory depression, and various other behavioural adverse effects that often result in patient non-compliance. In addition, the actual mechanisms of opioid-induced adverse reactions over long-term use are not entirely understood.
- opioids
- morphine
- analgesia
- adverse effects
1. Introduction
1.1. Pain
According to the International Association for the Study of Pain (IASP), pain is defined as an unpleasant sensory and emotional experience associated with actual potential tissue damage or described in terms of such damage [1]. Pain can be categorised as nociceptive, neuropathic or nociplastic pain (a combination of both that cannot be entirely explained as nociceptive or neuropathic). Nociceptive pain is generated as a warning signal transmitted to the brain about the possible damage of a non-neural tissue [1,2]. In contrast, neuropathic pain usually results from damage to neural tissue by a disease, toxin or infection [1,3]. The third type of pain, nociplastic pain, is a complex pain, not completely defined but probably caused by an alteration of neurons’ pain response and increased sensitivity of the central nervous system (CNS). Nociceplastic pain generally presents over 3 months of duration with symptoms such as hyperalgesia and regional pain sensations and is commonly observed in patients with cancer and other long-term chronic disorders [4–6]. The opioid system is a physiological control system that modulates pain, emotions, immune defence and various other physiological responses. The opioid system involves the communication and coordination of a significant number of endogenous opioid peptides and several types of opioid receptors in the CNS and peripheral nervous system. This system also significantly modulates numerous sensory, emotional, motivational and cognitive functions, as well as addictive behaviours [7,8,9]. It is also involved in other physiological functions, including responses to stress, respiration, gastrointestinal transit, endocrine and immune functions [10]. These responses are orchestrated by opioid ligands that bind to specific opioid receptors to induce analgesia and behavioural effects in vivo. Therefore, to understand the pharmacological effects of specific opioids, it is first essential to clarify the specific roles of each opioid receptor type.
According to the International Association for the Study of Pain (IASP), pain is defined as an unpleasant sensory and emotional experience associated with actual potential tissue damage or described in terms of such damage [1]. Pain can be categorised as nociceptive, neuropathic or nociplastic pain (a combination of both that cannot be entirely explained as nociceptive or neuropathic). Nociceptive pain is generated as a warning signal transmitted to the brain about the possible damage of a non-neural tissue [1][2]. In contrast, neuropathic pain usually results from damage to neural tissue by a disease, toxin or infection [1][3]. The third type of pain, the nociplastic pain, is a complex pain, not completely defined but probably caused by an alteration of neurons’ pain response and an increased sensitivity of the central nervous system (CNS). Nociceplastic pain generally presents over 3 months of duration with symptoms such as hyperalgesia and regional pain sensations and is commonly observed in patients with cancer and other long-term chronic disorders [4][5][6]. The opioid system is a physiological control system that modulates pain, emotions, immune defence and various other physiological responses. The opioid system involves the communication and coordination of a significant number of endogenous opioid peptides and several types of opioid receptors in the CNS and peripheral nervous system. This system also significantly modulates numerous sensory, emotional, motivational and cognitive functions, as well as addictive behaviours [7][8][9]. It is also involved in other physiological functions, including responses to stress, respiration, gastrointestinal transit, endocrine and immune functions [10]. These responses are orchestrated by opioid ligands that bind to specific opioid receptors to induce analgesia and behavioural effects in vivo. Therefore, to understand the pharmacological effects of specific opioids, it is first essential to clarify the specific roles of each opioid receptor type.
1.2. Opioid Receptors
The presence of opioid receptors was first proposed in 1954 [11]. However, the first evidence of the multiplicity of opioid receptors was only described in 1976 [12]. According to the International Union of Basic and Clinical Pharmacology (IUPHAR) and the British Pharmacological Society (BPS) joint IUPHAR/BPS Guide to Pharmacology, opioid receptors are classified into μ (Mu: MOP), δ (delta: DOP) and κ (kappa: KOP) receptors, as well as the non-classical nociception (NOP) receptor [13,14].
Opioid receptors belong to the family of seven-transmembrane helical G protein-coupled receptors (GPCRs) and share about 60% homology in the amino acid composition. These receptors display an extracellular N-terminus and an intracellular C-terminus and are coupled with heterotrimeric Gi/Go proteins [15,16,17][13][14][15]. Opioid ligands bind to opioid receptors by establishing ligand–receptor interactions in the binding pockets of the receptor, which are situated in the transmembrane helices. The binding pocket of opioid receptors can be divided into two distinct regions; the lower part (intracellular side) of the receptor is highly conserved for opioids (non-specific ‘message’ region), and the higher part of the pocket (extracellular side) contains divergent residues that confer selectivity (‘address’ region) to opioid receptor types; binding also depends on the type of the opioid ligand [18,19][16][17]. In 2012, the first molecular structures of all four opioid receptors were described in several reports [18,20,21,22][16][18][19][20].
Although all types of opioid receptor types modulate analgesia, the MOP receptor is thought to be dominant for its pain-relieving effects [23,24,25,26][21][22][23][24]. The major limitation of targeting the MOP receptor for analgesia is that it is also responsible for the induction of tolerance [27][25] and other undesirable adverse effects including addiction [28[26][27],29], dependence, respiratory depression [30][28] and constipation [31][29]. The MOP receptor is expressed in the brain, spinal cord and elsewhere in the body, and the adverse effects are relevant to its site of activation [28][26]. For example, in the gut, MOP receptor activation can cause constipation. However, the most important activation site is in the brain, as the MOP receptor drives hedonic reward, reinforcing, addictive, tolerance, dependence and withdrawal symptoms [32][30]. It is presumed that peripherally restricted MOP receptor agonists (that do not pass the blood–brain barrier) mediate local analgesia (effective against inflammatory or neuropathic pain) with reduced centrally mediated adverse effects [28][26]. MOP receptor-related adverse events are of great clinical concern and justify the characterisation of other opioid receptor types as suitable drug targets to induce analgesia. Unfortunately, the other three opioid receptor types (DOP, KOR and NOP receptors) do not have the same efficacy in mediating analgesia compared to the MOP receptor. DOP receptor agonists are generally less effective to treat acute thermal pain compared to inflammatory [33[31][32][33],34,35], neuropathic [36,37][34][35] and cancer-associated bone pain [38][36]. SNC80 and deltorphin II, two selective DOP receptor agonists, show significant anti-hyperalgesic effects, but these agonists are less potent or less efficacious in inducing thermal antinociceptive effects [34][32]. In addition, the use of DOP receptor agonists is limited, since DOP receptor-induced analgesia appears to require the presence of a pro-inflammatory state [39,40][37][38]. While DOP receptor agonists only produce moderate analgesia in non-human primates [41[39][40][41],42,43], despite being effective in rodent models of chronic pain [44][42], they are associated with convulsions in mice [45][43] and non-human primates [41,42,43][39][40][41]. Additionally, KOP receptor agonists are reported to reduce visceral [46[44][45],47], inflammatory [48,49][46][47] and neuropathic pain [50[48][49],51], but they also produce CNS-associated adverse events (i.e., dysphoria, psychotomimesis) [52,53,54][50][51][52]. While selective KOP and DOP receptor agonists lack some of the MOP receptor-mediated liabilities, such as constipation, respiratory depression and addiction, they display a side effect profile of their own [55][53]. Several NOP receptor agonists are reported to have antinociceptive effects in rodent [56,57][54][55] and primate models [58,59,60][56][57][58] and are associated with a reduced risk for abuse [61][59]. However, systemic administration of NOP agonists did not produce spinal analgesia in rodents [62[60][61],63], while showing efficacy after intrathecal administration in primates and rodent models of neuropathic pain [57,58,61,64][55][56][59][62]. Overall, MOP receptor agonists, despite their adverse effects, remain the most efficacious drugs in providing pain relief and are thus widely used in the clinic [26,65][24][63].
In the investigation on the role of specific opioid receptors and their ligands in pain modulation, antinociceptive tolerance and adverse behavioural effects, the generation of knockout animals has provided significant knowledge on the in vivo physiological role of the opioid system. For example, in MOP receptor knockout mice, MOP receptor agonist-induced antinociception and their associated side effects (e.g., hyperlocomotion, respiratory depression, inhibition of gastrointestinal tract transit, reward and withdrawal effects) were effectively abolished [23,66,67][21][64][65]. At the same time, morphine efficiently induced analgesia in DOP [68][66] and KOP [69][67] receptor knockout mice, albeit with reduced adverse effects (i.e., tolerance and withdrawal response). Similarly, KOP receptor agonists are also reported to induce analgesia in MOP [70][68] and DOP [68][66] receptor knockout mice, while predictably, in KOP receptor knockout animals, this effect was not observed [69][67]. However, DOP receptor agonists show only reduced levels of analgesia in DOP receptor knockout mice [68][66], although a mixed effect (decreased/maintained) on analgesia was observed in MOP receptor knockout mice [70,71][68][69].
1.3. Endogenous Opioid Ligands
Opioid ligands are from both endogenous and exogenous origins. Evidence of the existence of endogenous ligands for the opioid receptors was obtained in the 1970s, and the structures of [Leu]- and [Met]-enkephalin were reported in 1975 [72][70]. Endogenous opioid peptides are found in the CNS and peripheral nervous system and in the gastrointestinal tract [73][71]. These peptides are derived from the four different precursors pro-enkephalin, pro-dynorphin, pro-opiomelanocortin and prepro-nociceptin [15,74,75,76][13][72][73][74]. Pro-enkephalin contains two 267 amino acid polypeptides [77][75], and mainly produces the pentapeptides [Leu]- and [Met]-enkephalins [78,79][76][77] with selectivity for MOP and DOP receptors. Dynorphins are mainly big dynorphin, dynorphin A, dynorphin B and α-neo-endorphin and interact mainly with the KOP receptor [80,81][78][79]. Endorphins are derived from pro-opiomelanocortin [82][80] and are expressed as α-, β- and γ-endorphins [83][81]. While endorphins activate the MOP receptor, the prepro-nociceptin-derived neuropeptide nociceptin/orphanin FQ binds to the NOP receptor [7]. Endogenous opioids affect a multitude of physiological functions, such as pain modulation and analgesia, stress and emotional responses, tolerance and dependence, learning and memory, addiction, sexual activity and control of hormone levels, neurological disorders, eating and drinking behaviour, gastrointestinal, renal and hepatic functions, cardiovascular responses, respiration, thermoregulation and immunological responses [84,85][82][83].
1.4. Exogenous Opioid Ligands
Over more than 8000 years, the poppy plant (Papaver somniferum) and the opioids derived from it have been used for pain relief. In a Sumerian ideogram, the poppy plant was known as a “plant of joy” [85][83]. Crude opium admixtures were widely used in different British and German medicines in the 16th century, and effects like pain tolerance and physical dependence on opioids were noted at this time. In 1805, Friedrich Sertürner isolated morphium (morphine) and named it after the Greek god Morpheus (the “God of sleep and dreams”). Within two decades after the initial isolation of morphine, commercial production of morphine started, and morphine became available on the European market. Subsequently, after the invention of hypodermic syringes in the middle of the 19th century, morphine was injected systematically into painful areas [85][83].
Currently, different alkaloids extracted from the poppy plant (Papaver somniferum), including opium, morphine and codeine, are still used for pain relief, mood disorders and palliative care. In addition, several semi-synthetic and synthetic opioids, such as buprenorphine, dextropropoxyphene, hydromorphone, oxycodone, pethidine, fentanyl, methadone, tapentadol and tramadol, are widely used in patients that suffer from surgical or chronic pain [86][84].
2. Opioid-Induced Adverse Effects
2.1. Analgesic Tolerance
The development of analgesic tolerance to opioids after repetitive administration is one of the major limitations for their chronic use in the clinic. Morphine is one of the most effective and widely prescribed drugs against severe pain [26,119][24][85]. However, long-term morphine treatment is discouraged in the clinic due to the risk of adverse effects, including analgesic tolerance [120,121][86][87]. Tolerance manifests as decreased drug efficacy following repeated administration [122][88]. Therefore, to maintain efficacy, dose increments are required, which in turn contribute to generating cellular desensitisation, tolerance, physical dependence and behavioural withdrawal symptoms. In addition, increased morphine dosing is frequently required due to amplified disease progression rather than analgesic tolerance [123][89].
The clinical management of analgesic tolerance involves opioid rotation and the combination of opioids with adjuvants [124,125,126,127][90][91][92][93]. Adjuvants, such as gabapentin, pregabalin, dexamethasone, naproxen, ibuprofen, carbamazepine, aspirin, venlafaxine and acetaminophen, are combined with opioid analgesics in patients that require long-term analgesic treatment [128,129][94][95]. Similarly, in preclinical studies, a combination of opioids and non-opioid adjuvants or combinations of opioid agonist and antagonist are used to prevent antinociceptive tolerance [130,131,132,133][96][97][98][99]. The activation of the opioid receptors leads to receptor phosphorylation by GPCR kinases, which promotes the interaction with β-arr [134,135][100][101]. Both phosphorylation and interaction with β-arr are required for subsequent receptor internalisation [134,135][100][101]. This internalised receptor can be proteolytically degraded. However, receptors can also be recycled in endosomes to be returned to the cell membrane [135,136][101][102]. This process is called receptor trafficking. In addition, de novo receptor synthesis ensures that new opioid receptors are produced and transported to the cell membrane via the trans-Golgi network [135][101]. Prolonged treatment with opioids increases the number of inactive (phosphorylated) receptors on the membrane, as well as the number of de novo synthesised receptors [135,136][101][102]. A clinical study on the use of opioids in cancer-related chronic pain showed that chronic administration of opioids induces increased methylation of the MOP receptor gene (OPRM1) on peripheral leucocytes and causes analgesic tolerance, but the article reported that a preclinical study on mice showed that targeted re-expression of the MOP receptor (by gene therapy) in cancer cells can reverse analgesic tolerance [137][103].
Specifically, chronic exposure to morphine leads to the selective recruitment of β-arr2 but not of β-arr1 [138][104]. In contrast to the interaction with β-arr1, which leads to receptor recycling, β-arr2 does not results in opioid receptor recycling but increases the number of inactive receptors on the cell membrane. This process is associated with insufficient analgesia [138][104]. Although the molecular mechanisms that lead to opioid tolerance are not entirely clear, both desensitisation and trafficking are assumed to be the key factors that lead to insufficient analgesia [107,135,138,139][105][101][104][106]. Although it is essential to delineate the exact molecular mechanisms resulting in opioid tolerance [138[104][107][108][109][110],140,141,142,143], it is also important to understand how chronic morphine dosing itself can influence analgesic tolerance and associated behavioural dependence [144,145][111][112].
The antinociceptive effects of morphine and other opioids in preclinical studies are commonly measured as central (brain and spinal cord) or peripheral antinociception [146,147][113][114]. The commonly used tail-flick test potentially measures spinal-mediated nociception, while the hot-plate assay largely measures supraspinal-mediated nociception [147,148][114][115]. Generally, one such antinociception test is performed in preclinical studies with repeated morphine treatment. As a result, the progression of antinociceptive tolerance measured by a single pain assay may be different when using another assay [145][112].
2.2. Addiction and Physical Dependence
Apart from analgesic tolerance, long-term opioid treatment also causes behavioural adverse effects like physical dependence and addiction to these drugs. Physical dependence consists of craving for a drug either for pleasure or to avoid the occurrence of withdrawal symptoms following a reduction of the treatment dose or the intake of an opioid receptor antagonist [149,150][116][117]. Addiction indicates a loss of control of opioids use [150][117]. Physical dependence is associated with the upregulation of cAMP and noradrenergic signalling in the locus coeruleus (LC) neurons of the dorsal pontine tegmentum of the brainstem [151,152,153][118][119][120]. The molecular mechanism that initiates physical dependence and reward is associated with repeated opioid treatment [151,154,155][118][121][122]. Briefly, MOP receptor binding with opioids, like morphine, causes dopamine release by dopaminergic neurons in the VTA, VTA neurons transfer dopamine to the NAc, and this induces a pleasure feeling [154][121] (Figure 1). After chronic intake of opioids, a larger amount of opioids is required gradually to stimulate the VTA neurons and sustain the release of a similar amount of dopamine in the NAc. Thus, patients become dependent and tend to take more drugs to feel better [154][121]. The LC region of the brain that controls noradrenaline release is responsible for the dependence and reward processes [154][121].
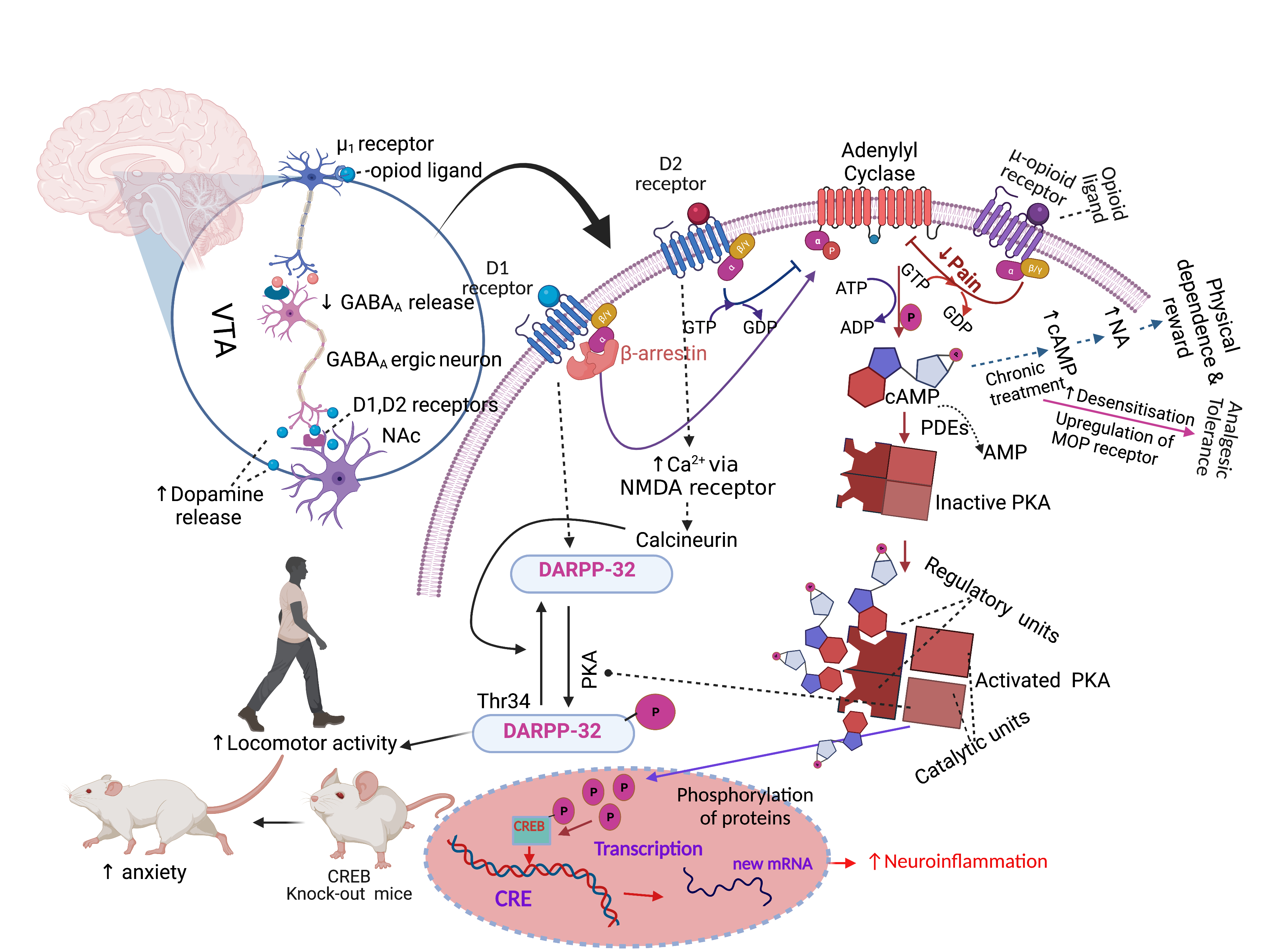
Figure 1. Opioid-induced analgesic and behavioural effects. Abbreviations: GABA, gamma-aminobutyric; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CRE, cAMP-responsive element; CREB, cAMP-responsive element binding protein; D1, dopamine receptor-1; DARPP-32, dopamine- and cAMP-regulated phosphoprotein of 32 kDa; NMDA, N-methyl-D-aspartate receptor; PDEs, Cyclic nucleotide phosphodiesterases; NA, noradrenaline, VTA, ventral tegmental area; NAc, nucleus accumbens. Symbols: solid arrow, strong activation; dashed arrow, moderate activation; solid T-shaped line, strong inhibition; dashed T-shaped line, moderate inhibition, upward arrow, increased effect; downward arrow, decreased effect. The figure was made with www.biorender.com (accessed on 16 November 2021) and adapted from 10.3390/ph14111091.
Endogenous opioids bind to opioid receptors in neuronal cell bodies of the LC and stimulate adenylyl cyclase to convert ATP to cAMP, but acute opioid intake, e.g., of morphine or heroine, inhibits the conversion, and as a result, less cAMP is produced. As noradrenaline release is stimulated by cAMP, less noradrenaline is released in the LC [154][121]. Noradrenaline stimulates wakefulness, respiration, and several other processes. Repeated opioids intake causes desensitisation of the opioid receptors; thus, neuronal cells produce a similar amount of ATP and cAMP in the presence of a higher concentration of opioids in the LC [154][121]. Chronic morphine administration increases the levels of type I and VII of adenylyl cyclase, PKA subunits and several phosphoproteins (e.g., CREB) and results in the hyperactivation of the cAMP pathway [152][119] (Figure 1). Then, if the patient stops taking opioids, this causes a massive release of noradrenaline in the LC neurons, which causes anxiety, nervousness and muscle cramps [154][121]. Clinical guidelines for long-term opioid use propose a “start low and go slow” dosing regimen to prevent addiction, physical dependence, overdosing or abuse [120,156,157,158,159,160][86][123][124][125][126][127]. Therefore, clinical guidelines propose administering the smallest effective dose [161][128], rather than aiming for adequate long-term pain relief [162][129].
2.3. Constipation
Constipation is a very common unwanted side effect of opioids and is caused by the activation of the MOP receptor in the enteric nervous system [163,164][130][131]. Opioids bind to MOP receptors in enteric neurons and delay gastrointestinal (GI) transit time, which also stimulates non-propulsive GI motility and pylorus and ileocecal sphincters [134][100]. Morphine treatment increases the expression of aquaporin-3 (AQP3) water channels in the colon by increased secretion of serotonin (5-HT), which increases water absorption from the luminal part to the vascular part of the colon [165][132]. As a result, constipation develops by increased fluid absorption from the large intestine along with less electrolyte secretion by the intestinal lumen [164][131]. In contrast, chronic morphine treatment does not produce tolerance to reduced GI motility in the lower GI tract, while it induces analgesic tolerance and leaves GI motility unaffected in the upper GI tract [166][133]. As a result, patients over a long-term opioid treatment continuously suffer from constipation. Constipation affects about 40% of patients with chronic oral opioid treatment, and therefore, different laxatives and non-medication approaches (e.g., fibrous diet, hydration) are used to provide comfort to the patients [149,167,168,169][116][134][135][136]. In addition, opioids combined with a low dose of opioid antagonists, such as naloxone, methylnaltrexone or alvimopan, are effective in reducing constipation without affecting pain relief and induce fewer withdrawal symptoms [169,170,171,172][136][137][138][139].
2.4. Respiratory Depression
Respiratory depression occurs less frequently compared to other adverse effects, but typically, it can have fatal consequences [149,181][116][140]. Similar to the other side effects, opioid-induced respiratory depression is mediated by the MOP receptor [182,183,184][141][142][143]. For example, fentanyl does not induce respiratory depression in MOP receptor knockout mice, indicating that the MOP receptor is responsible for respiratory depression [185][144]. Neurons of the pre-Bötzinger complex, a sub-region of the ventrolateral medulla, are responsible for controlling autonomic neuronal functions, including normal respiration [184][143]. The neurons of the pre-Bötzinger complex express a variety of receptors including neurokinin-1, serotonin (5-HT) and MOP receptors [184][143]. Inhibition of neurons that generate respiratory rhythms in the pre-Bötzinger complex cause respiratory depression [186][145]. MOP receptor activation inhibits adenylyl cyclase and reduces the synthesis of intracellular cAMP, which is thought to depress the respiratory neurons, as reduced cAMP levels in the cytoplasm reduce neuronal excitability by an unknown mechanism [134][100]. On the other hand, serotonin receptors in this region stimulate respiration [186,187][145][146]. The 5-HT1(a) receptors are expressed widely on respiratory neurons and are stimulated by reduced cAMP levels that activate the glycine receptor type α3 (GlyRα3) [188][147]. The activated GlyRα3 receptor inhibits neurons contributing to respiratory depression. This effect is independent of the MOP receptor-induced signal transduction pathway [188][147]. Therefore, multiple non-opioid receptors together with the MOP receptor are involved in the control of respiration and opioid-induced respiratory depression. Although high-dose opioid users are at risk of respiratory depression [189][148], a selective peripherally selective opioid antagonist can effectively reduce the incidence of respiratory depression without significant withdrawal symptoms [190][149].
2.5. Other Adverse Effects
In addition to analgesic tolerance, physical dependence and addiction as major adverse effects of long-term opioid treatment, this discussion also needs to address other behavioural side effects observed in the clinic [191][150]. Morphine-induced biphasic behavioural effects are well known from preclinical studies and include initial motor suppression and subsequent hyper-excitation [192,193,194,195,196,197,198][151][152][153][154][155][156][157]. An open-field arena is widely used to assess motor behaviour and typically includes horizontal movement, rearing (vertical movement) and turning behaviour. Morphine-induced horizontal locomotion, turning and circling behaviours are related to the dopaminergic system [195,199,200,201][154][158][159][160]. Morphine treatment induces the dopamine receptor-1 (D1)-dependent βarr-2/phospho-ERK (βarr2/pERK) signalling complex, which stimulates morphine-induced horizontal locomotion. However, the effects were absent in D1 and D2 receptor knockout mice [202][161]. Acute morphine administration induces phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), which activates the D1 receptor on dopaminergic neurons of NAc, substantia nigra and dorsal striatum and stimulates locomotor activity [202,203,204,205][161][162][163][164]. The D1, D2 and D3 receptors are responsible for the control of locomotion, learning and memory-related functions [206][165]. Opioid-induced activation of μ1-opioid receptor decreases gamma-aminobutyric (GABA) release in co-localised GABAA receptors in the ventral tegmental area (VTA), but stimulates dopamine release in the NAc, as the dopaminergic neurons in the NAc emerge from the VTA [207][166] (Figure 1). The VTA, NAc (ventral striatum) and substantia nigra dopaminergic system is also involved in motivation-, reward- and addiction-related behaviour [208,209,210,211][167][168][169][170]. Noticeably, the effects of chronic morphine treatment on cAMP/PKA/DARPP-32 signalling are not fully understood at present. However, rearing behaviour can indicate increased exploration and reduced anxiety, which are related to GABA inhibitory neurotransmission [212,213,214][171][172][173]. Activation of the CREB transcription factor regulates anxiety-related behaviours, as CREB-deficient mice show an increased anxiogenic response [215][174]. However, the behavioural changes in response to chronic morphine treatment are independent of MOP receptor, cyclin-dependent kinase 5 (cdk5) or adenylyl cyclase activities in relevant areas of the brain [216][175]. Studies suggested that the morphine-induced behavioural effects are probably derived from its binding to the KOP receptor but not to the MOP receptor [217,218][176][177]. The likely multiple mechanisms that link chronic morphine treatment to its behavioural effects are not completely understood but may be controlled by a combination of dopaminergic, GABAergic, opioidergic and additional unknown neuronal signals [107,202,203,204,212,213,214][105][161][162][163][171][172][173]. The combination of multiple independent behavioural measurements is generally regarded as the most reliable approach to assess the total motor effects induced by opioids [193,194,219,220,221][152][153][178][179][180].
References
- IASP. Pain, IASP Terminology. International Association for the Study of Pain (IASP). Available online: https://www.iasp-pain.org/resources/terminology/#pain (accessed on 26 October 2021).
- Woolf, C.J.; Bennett, G.J.; Doherty, M.; Dubner, R.; Kidd, B.; Koltzenburg, M.; Lipton, R.; Loeser, J.D.; Payne, R.; Torebjork, E. Towards a mechanism-based classification of pain? Pain 1998, 77, 227–229.
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964.
- Fitzcharles, M.-A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110.
- Nijs, J.; Lahousse, A.; Kapreli, E.; Bilika, P.; Saraçoğlu, I.; Malfliet, A.; Coppieters, I.; De Baets, L.; Leysen, L.; Roose, E.; et al. Nociplastic pain criteria or recognition of central sensitization? Pain phenotyping in the past, present and future. J. Clin. Med. 2021, 10, 3203.
- IASP. Central Sensitization, IASP Terminology. Available online: https://www.iasp-pain.org/resources/terminology/?navItemNumber=576#Centralsensitization (accessed on 25 August 2021).
- Cai, Z.; Ratka, A. Opioid system and Alzheimer’s disease. Neuromol. Med. 2012, 14, 91–111.
- Melzack, R. From the gate to the neuromatrix. Pain 1999, 82, S121–S126.
- Holden, J.E.; Jeong, Y.; Forrest, J.M. The endogenous opioid system and clinical pain management. AACN Clin. Issues Adv. Pr. Acute Crit. Care 2005, 16, 291–301.
- Le Merrer, J.; Becker, J.A.; Befort, K.; Kieffer, B.L. Reward processing by the opioid system in the brain. Physiol. Rev. 2009, 89, 1379–1412.
- Beckett, A.H.; Casy, A.F. Synthetic analgesics: Stereochemical considerations. J. Pharm. Pharmacol. 2011, 6, 986–1001.
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine-and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532.
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M.J.; Civelli, O.; Connor, M.; Cox, B.M.; Devi, L.A.; Evans, C.; et al. Opioid Receptors, Introduction. IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetoimmunopharmacology.org/GRAC/FamilyIntroductionForward?familyId=50 (accessed on 26 October 2021).
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990.
- Chen, Y.; Mestek, A.; Liu, J.; Yu, L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem. J. 1993, 295, 625–628.
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.-L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.-C. ORL1, a novel member of the opioid receptor family. FEBS Lett. 1994, 341, 33–38.
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nat. Cell Biol. 2012, 485, 400–404.
- Filizola, M.; Devi, L.A. How opioid drugs bind to receptors. Nat. Cell Biol. 2012, 485, 314–317.
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nat. Cell Biol. 2012, 485, 321–326.
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.-P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nat. Cell Biol. 2012, 485, 395–399.
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.-P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nat. Cell Biol. 2012, 485, 327–332.
- Matthes, H.W.D.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the µ-opioid-receptor gene. Nat. Cell Biol. 1996, 383, 819–823.
- Kieffer, B.L. Opioids: First lessons from knockout mice. Trends Pharmacol. Sci. 1999, 20, 19–26.
- Sora, I.; Takahashi, N.; Funada, M.; Ujike, H.; Revay, R.S.; Donovan, D.M.; Miner, L.L.; Uhl, G.R. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. USA 1997, 94, 1544–1549.
- Corbett, A.D.; Henderson, G.; McKnight, A.T.; Paterson, S.J. 75 years of opioid research: The exciting but vain quest for the Holy Grail. Br. J. Pharmacol. 2006, 147, S153–S162.
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; Von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of µ-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254.
- Machelska, H.; Celik, M.Ö. Advances in achieving opioid analgesia without side effects. Front. Pharmacol. 2018, 9, 1388.
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514.
- Romberg, R.; Sarton, E.; Teppema, L.; Matthes, H.W.; Kieffer, B.L.; Dahan, A. Comparison of morphine-6-glucuronide and morphine on respiratory depressant and antinociceptive responses in wild type and mu-opioid receptor deficient mice. Br. J. Anaesth. 2003, 91, 862–870.
- Mori, T.; Shibasaki, Y.; Matsumoto, K.; Shibasaki, M.; Hasegawa, M.; Wang, E.; Masukawa, D.; Yoshizawa, K.; Horie, S.; Suzuki, T. Mechanisms that underlie μ-opioid receptor agonist–induced constipation: Differential involvement of μ-opioid receptor sites and responsible regions. J. Pharmacol. Exp. Ther. 2013, 347, 91–99.
- Daniels, D.J.; Lenard, N.R.; Etienne, C.L.; Law, P.-Y.; Roerig, S.C.; Portoghese, P.S. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. USA 2005, 102, 19208–19213.
- Pradhan, A.A.A.; Walwyn, W.; Nozaki, C.; Filliol, D.; Erbs, E.; Matifas, A.; Evans, C.; Kieffer, B.L. Ligand-directed trafficking of the-opioid receptor in vivo: Two paths toward analgesic tolerance. J. Neurosci. 2010, 30, 16459–16468.
- Fraser, G.L.; Gaudreau, G.-A.; Clarke, P.B.S.; Ménard, D.P.; Perkins, M.N. Antihyperalgesic effects of δ opioid agonists in a rat model of chronic inflammation. Br. J. Pharmacol. 2000, 129, 1668–1672.
- Cahill, C.M.; Morinville, A.; Hoffert, C.; O’Donnell, D.; Beaudet, A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: Implications for pain control. Pain 2003, 101, 199–208.
- Gaveriaux-Ruff, C.; Nozaki, C.; Nadal, X.; Hever, X.C.; Weibel, R.; Matifas, A.; Reiss, D.; Filliol, D.; Nassar, M.A.; Wood, J.N.; et al. Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain 2011, 152, 1238–1248.
- Kabli, N.; Cahill, C.M. Anti-Allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 2007, 127, 84–93.
- Brainin-Mattos, J.; Smith, N.D.; Malkmus, S.; Rew, Y.; Goodman, M.; Taulane, J.; Yaksh, T.L. Cancer-related bone pain is attenuated by a systemically available δ-opioid receptor agonist. Pain 2006, 122, 174–181.
- Vanderah, T.W. Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26, S10–S15.
- Gendron, L.; Mittal, N.; Beaudry, H.; Walwyn, W. Recent advances on the δ opioid receptor: From trafficking to function. Br. J. Pharmacol. 2015, 172, 403–419.
- Dykstra, L.A.; Schoenbaum, G.M.; Yarbrough, J.; McNutt, R.; Chang, K.J. A novel delta opioid agonist, BW373U86, in squirrel monkeys responding under a schedule of shock titration. J. Pharmacol. Exp. Ther. 1993, 267, 875–882.
- Negus, S.S.; Butelman, E.R.; Chang, K.J.; DeCosta, B.; Winger, G.; Woods, J.H. Behavioral effects of the systemically active delta opioid agonist BW373U86 in rhesus monkeys. J. Pharmacol. Exp. Ther. 1994, 270, 1025–1034.
- Negus, S.S.; Gatch, M.; Mello, N.K.; Zhang, X.; Rice, K. Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J. Pharmacol. Exp. Ther. 1998, 286, 362–375.
- Nozaki, C.; Le Bourdonnec, B.; Reiss, D.; Windh, R.T.; Little, P.J.; Dolle, R.E.; Kieffer, B.L.; Gaveriaux-Ruff, C. δ-Opioid mechanisms for ADL5747 and ADL5859 effects in mice: Analgesia, locomotion, and receptor internalization. J. Pharmacol. Exp. Ther. 2012, 342, 799–807.
- Broom, D.C.; Nitsche, J.F.; Pintar, J.E.; Rice, K.C.; Woods, J.H.; Traynor, J.R. Comparison of receptor mechanisms and efficacy requirements for δ-agonist-induced convulsive activity and antinociception in mice. J. Pharmacol. Exp. Ther. 2002, 303, 723–729.
- Eisenach, J.C.; Carpenter, R.; Curry, R. Analgesia from a peripherally active κ-opioid receptor agonist in patients with chronic pancreatitis. Pain 2003, 101, 89–95.
- Riviere, P. Peripheral kappa-opioid agonists for visceral pain. Br. J. Pharmacol. 2004, 141, 1331–1334.
- Moon, S.W.; Park, E.H.; Suh, H.R.; Ko, D.H.; Kim, Y.I.; Han, H.C. The contribution of activated peripheral kappa opioid receptors (kORs) in the inflamed knee joint to anti-nociception. Brain Res. 2016, 1648, 11–18.
- Bileviciute-Ljungar, I.; Spetea, M. Contralateral but not systemic administration of the κ-opioid agonist U-50,488H induces anti-nociception in acute hindpaw inflammation in rats. Br. J. Pharmacol. 2001, 132, 252–258.
- Keita, H.; Kayser, V.; Guilbaud, G. Antinociceptive effect of a kappa-opioid receptor agonist that minimally crosses the blood-brain barrier (ICI 204448) in a rat model of mononeuropathy. Eur. J. Pharmacol. 1995, 277, 275–280.
- Bileviciute-Ljungar, I.; Spetea, M. Contralateral, ipsilateral and bilateral treatments with the κ-opioid receptor agonist U-50,488H in mononeuropathic rats. Eur. J. Pharmacol. 2004, 494, 139–146.
- Dykstra, L.A.; Gmerek, D.E.; Winger, G.; Woods, J.H. Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Ther. 1987, 242, 413–420.
- Butelman, E.R.; Harris, T.J.; Kreek, M.-J. Effects of E-2078, a stable dynorphin A(1-8) analog, on sedation and serum prolactin levels in rhesus monkeys. Psychopharmacology 1999, 147, 73–80.
- Ko, M.C.H.; Johnson, M.D.; Butelman, E.R.; Willmont, K.J.; Mosberg, H.I.; Woods, J.H. Intracisternal nor-binaltorphimine distinguishes central and peripheral kappa-opioid antinociception in rhesus monkeys. J. Pharmacol. Exp. Ther. 1999, 291, 1113–1120.
- Peng, X.; Neumeyer, J.L. Kappa receptor bivalent ligands. Curr. Top. Med. Chem. 2007, 7, 363–373.
- Calo’, G.; Rizzi, A.; Cifani, C.; Di Bonaventura, M.V.M.; Regoli, D.; Massi, M.; Salvadori, S.; Lambert, D.G.; Guerrini, R. UFP-112 a potent and long-lasting agonist selective for the nociceptin/orphanin FQ receptor. CNS Neurosci. Ther. 2010, 17, 178–198.
- Rizzi, A.; Spagnolo, B.; Wainford, R.; Fischetti, C.; Guerrini, R.; Marzola, G.; Baldisserotto, A.; Salvadori, S.; Regoli, D.; Kapusta, D.R.; et al. In vitro and in vivo studies on UFP-112, a novel potent and long lasting agonist selective for the nociceptin/orphanin FQ receptor. Peptides 2007, 28, 1240–1251.
- Hu, E.; Calò, G.; Guerrini, R.; Ko, M.-C. Long-lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain 2010, 148, 107–113.
- Ko, M.-C.; Woods, J.H.; Fantegrossi, W.E.; Galuska, C.M.; Wichmann, J.; Prinssen, E.P. Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology 2009, 34, 2088–2096.
- Varty, G.B.; Lu, S.X.; Morgan, C.A.; Cohen-Williams, M.E.; Hodgson, R.A.; Smith-Torhan, A.; Zhang, H.; Fawzi, A.B.; Graziano, M.P.; Ho, G.D.; et al. The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8--3-phenyl-8-azabicyclooctan-3-ol (SCH 221510). J. Pharmacol. Exp. Ther. 2008, 326, 672–682.
- Lin, A.P.; Ko, M.-C. The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem. Neurosci. 2012, 4, 214–224.
- Dautzenberg, F.M.; Wichmann, J.; Higelin, J.; Py-Lang, G.; Kratzeisen, C.; Malherbe, P.; Kilpatrick, G.; Jenck, F. Pharmacological characterization of the novel nonpeptide orphanin FQ/nociceptin receptor agonist Ro 64-6198: Rapid and reversible desensitization of the ORL1 receptor in vitro and lack of tolerance in vivo. J. Pharmacol. Exp. Ther. 2001, 298, 812–819.
- Reiss, D.; Wichmann, J.; Tekeshima, H.; Kieffer, B.L.; Ouagazzal, A.-M. Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64-6198, on reactivity to acute pain in mice: Comparison to morphine. Eur. J. Pharmacol. 2008, 579, 141–148.
- Obara, I.; Przewlocki, R.; Przewlocka, B. Spinal and local peripheral antiallodynic activity of Ro64-6198 in neuropathic pain in the rat. Pain 2005, 116, 17–25.
- Pathan, H.; Williams, J. Basic opioid pharmacology: An update. Br. J. Pain 2012, 6, 11–16.
- Schuller, A.G.; King, M.A.; Zhang, J.; Bolan, E.; Pan, Y.; Morgan, D.; Chang, A.; Czick, M.E.; Unterwald, E.M.; Pasternak, G.W.; et al. Retention of heroin and morphine–6β–glucuronide analgesia in a new line of mice lacking exon 1 of MOR–1. Nat. Neurosci. 1999, 2, 151–156.
- Loh, H.H.; Liu, H.C.; Cavalli, A.; Yang, W.; Chen, Y.F.; Wei, L.N. mu Opioid receptor knockout in mice: Effects on ligand-induced analgesia and morphine lethality. Brain Res. Mol. Brain Res. 1998, 54, 321–326.
- Zhu, Y.; King, M.A.; Schuller, A.G.; Nitsche, J.F.; Reidl, M.; Elde, R.P.; Unterwald, E.; Pasternak, G.W.; Pintar, J.E. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in δ opioid receptor knockout mice. Neuron 1999, 24, 243–252.
- Simonin, F.; Valverde, O.; Smadja, C.; Slowe, S.; Kitchen, I.; Dierich, A.; Le Meur, M.; Roques, B.P.; Maldonado, R.; Kieffer, B.L. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 1998, 17, 886–897.
- Matthes, H.W.; Smadja, C.; Valverde, O.; Vonesch, J.L.; Foutz, A.S.; Boudinot, E.; Denavit-Saubie, M.; Severini, C.; Negri, L.; Roques, B.P.; et al. Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. J. Neurosci. 1998, 18, 7285–7295.
- Hosohata, Y.; Vanderah, T.W.; Burkey, T.H.; Ossipov, M.H.; Kovelowski, C.J.; Sora, I.; Uhl, G.R.; Zhang, X.; Rice, K.C.; Roeske, W.R.; et al. Delta-opioid receptor agonists produce antinociception and GTPgammaS binding in mu receptor knockout mice. Eur. J. Pharmacol. 2000, 388, 241–248.
- Lefebvre, R. Opioid peptides and their receptors. In Comparative Veterinary Pharmacology, Toxicology and Theraphy; Van Miert, A.S.J.P.A.M., Bogaert, M.G., Debackere, M., Eds.; Springer: Berlin/Heidelberg, Germany, 1986; pp. 447–453.
- Bergström, J.; Ahmed, M.; Li, J.; Ahmad, T.; Kreicbergs, A.; Spetea, M. Opioid peptides and receptors in joint tissues: Study in the rat. J. Orthop. Res. 2006, 24, 1193–1199.
- Callahan, P.; Pasternak, G.W. Opiates, opioid peptides, and their receptors. J. Cardiothorac. Anesth. 1987, 1, 569–576.
- Chaturvedi, K. Opioid peptides, opioid receptors and mechanism of down regulation. Indian J. Exp. Boil. 2003, 41, 5–13.
- Snyder, S.H.; Childers, S.R. Opiate receptors and opioid peptides. Annu. Rev. Neurosci. 1979, 2, 35–64.
- Marino, R.; Struck, J.; Hartmann, O.; Maisel, A.S.; Rehfeldt, M.; Magrini, L.; Melander, O.; Bergmann, A.; Di Somma, S. Diagnostic and short-term prognostic utility of plasma pro-enkephalin (pro-ENK) for acute kidney injury in patients admitted with sepsis in the emergency department. J. Nephrol. 2015, 28, 717–724.
- Roberts, E.; Shoureshi, P.; Kozak, K.; Szynskie, L.; Baron, A.; Lecaude, S.; Dores, R.M. Tracking the evolution of the proenkephalin gene in tetrapods. Gen. Comp. Endocrinol. 2007, 153, 189–197.
- Gonzalez Nunez, V.; Gonzalez Sarmiento, R.; Rodriguez, R.E. Characterization of zebrafish proenkephalin reveals novel opioid sequences. Brain Res. Mol. Brain Res. 2003, 114, 31–39.
- Merg, F.; Filliol, D.; Usynin, I.; Bazov, I.; Bark, N.; Hurd, Y.L.; Yakovleva, T.; Kieffer, B.L.; Bakalkin, G. Big dynorphin as a putative endogenous ligand for the kappa-opioid receptor. J. Neurochem. 2006, 97, 292–301.
- Gein, S. Dynorphins in regulation of immune system functions. Biochemistry 2014, 79, 397–405.
- Diamant, M.; Henricks, P.A.; Nijkamp, F.P.; de Wied, D. Beta-endorphin and related peptides suppress phorbol myristate acetate-induced respiratory burst in human polymorphonuclear leukocytes. Life Sci. 1989, 45, 1537–1545.
- Lolait, S.J.; Clements, J.A.; Markwick, A.J.; Cheng, C.; McNally, M.; Smith, I.; Funder, J.W. Pro-opiomelanocortin messenger ribonucleic acid and posttranslational processing of beta endorphin in spleen macrophages. J. Clin. Investig. 1986, 77, 1776–1779.
- Bodnar, R.J. Endogenous opiates and behavior: 2012. Peptides 2013, 50, 55–95.
- Fine, P.G.; Portenoy, R.K. A Clinical Guide to Opioid Analgesia; The McGraw-Hill Companies: New York, NY, USA, 2004; pp. 1–7.
- AMH. Analgesics (Ch 3). In Australian Medicines Handbook (AMH). Available online: https://amhonline.amh.net.au/chapters/chap-03?menu=vertical (accessed on 22 October 2017).
- Devereaux, A.L.; Mercer, S.L.; Cunningham, C.W. DARK classics in chemical neuroscience: Morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407.
- TG. Principles of Nonsteroidal Anti-Inflammatory Drug Use for Musculoskeletal Conditions in Adults. In eTG Complete Melbourne: Therapeutic Guidelines Limited. Available online: https://tgldcdp.tg.org.au/index (accessed on 29 December 2020).
- CDC. Prescription Opioids: Side Effects. Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/drugoverdose/opioids/prescribed.html (accessed on 29 December 2020).
- WHO. Guidelines for the Psychosocially Assisted Pharmacological Treatment of Opioid Dependence. World Health Organization, Geneva. 2009. Available online: http://apps.who.int/iris/bitstream/10665/43948/1/9789241547543_eng.pdf (accessed on 26 October 2021).
- Collin, E.; Poulain, P.; Gauvain-Piquard, A.; Petit, G.; Pichard-Leandri, E. Is disease progression the major factor in morphine ‘tolerance’ in cancer pain treatment? Pain 1993, 55, 319–326.
- Freye, E.; Anderson-Hillemacher, A.; Ritzdorf, I.; Levy, J.V. Opioid rotation from high-dose morphine to transdermal buprenorphine (Transtec®) in chronic pain patients. Pain Pract. 2007, 7, 123–129.
- Rhondali, W.; Tremellat, F.; LeDoux, M.; Ciais, J.-F.; Bruera, E.; Filbet, M. Methadone rotation for cancer patients with refractory pain in a palliative care unit: An observational study. J. Palliat. Med. 2013, 16, 1382–1387.
- Leppert, W.; Kowalski, G. Long-term administration of high doses of transdermal buprenorphine in cancer patients with severe neuropathic pain. OncoTargets Ther. 2015, 8, 3621–3627.
- Sutou, I.; Nakatani, T.; Hashimoto, T.; Saito, Y. Fentanyl Tolerance in the Treatment of Cancer Pain: A Case of Successful Opioid Switching from Fentanyl to Oxycodone at a Reduced Equivalent Dose. J. Pain Palliat. Care Pharmacother. 2015, 29, 161–165.
- Shinde, S.; Gordon, P.; Sharma, P.; Gross, J.; Davis, M.P. Use of non-opioid analgesics as adjuvants to opioid analgesia for cancer pain management in an inpatient palliative unit: Does this improve pain control and reduce opioid requirements? Support. Care Cancer 2014, 23, 695–703.
- Raptis, E.; Vadalouca, A.; Stavropoulou, E.; Argyra, E.; Melemeni, A.; Siafaka, I. Pregabalin Vs. Opioids for the treatment of neuropathic cancer pain: A prospective, head-to-head, randomized, open-label study. Pain Pract. 2013, 14, 32–42.
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.S.; Takemori, A.E. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303.
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of morphine tolerance and dependence with the highly selective delta-opioid receptor antagonist TIPP. Eur. J. Pharmacol. 1995, 286, 105–108.
- Hepburn, M.J.; Little, P.J.; Gingras, J.; Kuhn, C.M. Differential effects of naltrindole on morphine-induced tolerance and physical dependence in rats. J. Pharmacol. Exp. Ther. 1997, 281, 1350–1356.
- Roy, S.; Guo, X.; Kelschenbach, J.; Liu, Y.; Loh, H.H. In vivo activation of a mutant-opioid receptor by naltrexone produces a potent analgesic effect but no tolerance: Role of-Receptor activation and-receptor blockade in morphine tolerance. J. Neurosci. 2005, 25, 3229–3233.
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255.
- Bie, B.; Pan, Z.Z. Trafficking of central opioid receptors and descending pain inhibition. Mol. Pain 2007, 3, 37.
- Bs, A.C.B.; Whistler, J.L. How to design an opioid drug that causes reduced tolerance and dependence. Ann. Neurol. 2010, 67, 559–569.
- Viet, C.T.; Dang, D.; Aouizerat, B.E.; Miaskowski, C.; Ye, Y.; Viet, D.T.; Ono, K.; Schmidt, B.L. OPRM1 methylation contributes to opioid tolerance in cancer patients. J. Pain 2017, 18, 1046–1059.
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed Interactions with Specific β-Arrestins Determine μ-Opioid Receptor Trafficking, Ubiquitination, and Dephosphorylation. J. Biol. Chem. 2011, 286, 31731–31741.
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381.
- Arttamangkul, S.; Heinz, D.A.; Bunzow, J.R.; Song, X.; Williams, J.T. Cellular tolerance at the µ-opioid receptor is phosphorylation dependent. eLife 2018, 7, e34989.
- Martini, L.; Whistler, J.L. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007, 17, 556–564.
- Bourova, L.; Vosahlikova, M.; Kagan, D.; Dlouha, K.; Novotny, J.; Svoboda, P. Long-term adaptation to high doses of morphine causes desensitization of mu-OR- and delta-OR-stimulated G-protein response in forebrain cortex but does not decrease the amount of G-protein alpha subunits. Med. Sci. Monit. 2010, 16, 260–270.
- Gupta, A.; Mulder, J.; Gomes, I.; Rozenfeld, R.; Bushlin, I.; Ong, E.; Lim, M.; Maillet, E.; Junek, M.; Cahill, C.M. Increased abundance of opioid receptor heteromers following chronic morphine administration. Sci. Signal. 2010, 3, ra54.
- Leah, P.M.; Heath, E.M.L.; Balleine, B.W.; Christie, M. Chronic morphine reduces surface expression of δ-Opioid receptors in subregions of rostral striatum. Neurochem. Res. 2015, 41, 500–509.
- Paul, A.; Gueven, N.; Dietis, N. Profiling the effects of repetitive morphine administration on motor behavior in rats. Molecules 2021, 26, 4355.
- Paul, A.K.; Gueven, N.; Dietis, N. Morphine dosing strategy plays a key role in the generation and duration of the produced antinociceptive tolerance. Neuropharmacology 2017, 121, 158–166.
- McQuay, H. Central analgesics. Acta Neurochir. Suppl. 1987, 38, 41–43.
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652.
- Gårdmark, M.; Höglund, A.U.; Hammarlund-Udenaes, M. Aspects on Tail-Flick, Hot-Plate and electrical stimulation tests for morphine antinociception. Pharmacol. Toxicol. 1998, 83, 252–258.
- Labianca, R.; Sarzi-Puttini, P.; Zuccaro, S.M.; Cherubino, P.; Vellucci, R.; Fornasari, D. Adverse effects associated with non-opioid and opioid treatment in patients with chronic pain. Clin. Drug Investig. 2012, 32, 53–63.
- Juurlink, D.N.; Dhalla, I.A. Dependence and addiction during chronic opioid therapy. J. Med. Toxicol. 2012, 8, 393–399.
- Ballantyne, J.C.; LaForge, S.K. Opioid dependence and addiction during opioid treatment of chronic pain. Pain 2007, 129, 235–255.
- Nestler, E.J.; Aghajanian, G.K. Molecular and Cellular Basis of Addiction. Science 1997, 278, 58–63.
- Counts, S.E.; Mufson, E.J. Chapter 12-Locus coeruleus. In The Human Nervous System, 3rd ed.; Mai, J.K., Paxinos, G., Eds.; Academic Press: San Diego, CA, USA, 2012; pp. 425–438.
- Kosten, T.R.; George, T.P. The neurobiology of opioid dependence: Implications for treatment. Sci. Pract. Perspect. 2002, 1, 13–20.
- Listos, J.; Łupina, M.; Talarek, S.; Mazur, A.; Orzelska-Górka, J.; Kotlińska, J. The mechanisms involved in morphine addiction: An overview. Int. J. Mol. Sci. 2019, 20, 4302.
- Dowell, D.; Haegerich, T.M.; Chou, R. CDC Guideline for prescribing opioids for chronic pain—United States, 2016. JAMA 2016, 315, 1624–1645.
- (PHE) Public Health England; Faculty of Medicine of the Royal College of Anaesthetists; Royal College of General Practitioners; The British Pain Society. Managing Persistent Pain in Secure Settings. 2013. Available online: http://www.nta.nhs.uk/uploads/persistentpain.pdf (accessed on 24 September 2016).
- NOUGG. Canadian Guideline for Safe and Effective Use of Opioids for Chronic Non-Cancer Pain. Canada. National Opioid Use Guideline Group (NOUGG). 2010. Available online: http://nationalpaincentre.mcmaster.ca/opioid/ (accessed on 24 September 2016).
- American Society of Anesthesiologists Task Force on Chronic Pain Management. American Society of Regional Anesthesia and Pain Medicine Practice Guidelines for Chronic Pain Management. Anesthesiology 2010, 112, 810–833.
- Trescot, A.M.; Helm, S.; Hansen, H.; Benyamin, R.; Glaser, S.E.; Adlaka, R.; Patel, S.; Manchikanti, L. Opioids in the management of chronic non-cancer pain: An update of American Society of the Interventional Pain Physicians’ (ASIPP) Guidelines. Pain Physician 2008, 11, S5–S62.
- Kirpalani, D. How to maximize patient safety when prescribing opioids. PM&R 2015, 7, S225–S235.
- Stein, C.; Reinecke, H.; Sorgatz, H. Opioid use in chronic noncancer pain: Guidelines revisited. Curr. Opin. Anaesthesiol. 2010, 23, 598–601.
- Streicher, J.M.; Bilsky, E.J. Peripherally acting μ-Opioid receptor antagonists for the treatment of opioid-related side effects: Mechanism of action and clinical implications. J. Pharm. Pr. 2017, 31, 658–669.
- Camilleri, M. Opioid-Induced constipation: Challenges and therapeutic opportunities. Am. J. Gastroenterol. 2011, 106, 835–842.
- Kon, R.; Ikarashi, N.; Hayakawa, A.; Haga, Y.; Fueki, A.; Kusunoki, Y.; Tajima, M.; Ochiai, W.; Machida, Y.; Sugiyama, K. Morphine-Induced constipation develops with increased aquaporin-3 expression in the colon via increased serotonin secretion. Toxicol. Sci. 2015, 145, 337–347.
- Akbarali, H.I.; Inkisar, A.; Dewey, W.L. Site and mechanism of morphine tolerance in the gastrointestinal tract. Neurogastroenterol. Motil. 2014, 26, 1361–1367.
- Swegle, J.M.; Logemann, C. Management of common opioid-induced adverse effects. Am. Fam. Physician 2006, 74, 1347–1354.
- Cherny, N.; Ripamonti, C.; Pereira, J.; Davis, C.; Fallon, M.; McQuay, H.; Mercadante, S.; Pasternak, G.; Ventafridda, V. For the expert working group of the european association of palliative care network strategies to manage the adverse effects of oral morphine: An evidence-based report. J. Clin. Oncol. 2001, 19, 2542–2554.
- Prichard, D.; Norton, C.; Bharucha, A.E. Management of opioid-induced constipation. Br. J. Nurs. 2016, 25, S4–S5, S8–S11.
- Meissner, W.; Schmidt, U.; Hartmann, M.; Kath, R.; Reinhart, K. Oral naloxone reverses opioid-associated constipation. Pain 2000, 84, 105–109.
- Shimoyama, N.; Shimoyama, M. Treatment of constipation in chronic pain patients. Masui. Jpn. J. Anesthesiol. 2013, 62, 822–828.
- Burness, C.B.; Keating, G.M. Oxycodone/naloxone prolonged-release: A review of its use in the management of chronic pain while counteracting opioid-induced constipation. Drugs 2014, 74, 353–375.
- Sultan, P.; Gutierrez, M.C.; Carvalho, B. Neuraxial morphine and respiratory depression. Drugs 2011, 71, 1807–1819.
- Kuo, A.; Wyse, B.D.; Meutermans, W.; Smith, M.T. In vivo profiling of seven common opioids for antinociception, constipation and respiratory depression: No two opioids have the same profile. Br. J. Pharmacol. 2014, 172, 532–548.
- Boom, M.; Niesters, M.; Sarton, E.; Aarts, L.; Smith, T.W.; Dahan, A. Non-analgesic effects of opioids: Opioid-induced respiratory depression. Curr. Pharm. Des. 2012, 18, 5994–6004.
- Kamei, J.; Ohsawa, M.; Hayashi, S.-S.; Nakanishi, Y. Effect of chronic pain on morphine-induced respiratory depression in mice. Neuroscience 2011, 174, 224–233.
- Hill, R.; Santhakumar, R.; Dewey, W.; Kelly, E.; Henderson, G. Fentanyl depression of respiration: Comparison with heroin and morphine. Br. J. Pharmacol. 2020, 177, 254–265.
- Manzke, T.; Guenther, U.; Ponimaskin, E.G.; Haller, M.; Dutschmann, M.; Schwarzacher, S.; Richter, D.W. 5-HT 4(a) Receptors avert opioid-induced breathing depression without loss of analgesia. Science 2003, 301, 226–229.
- Pattinson, K.T.S. Opioids and the control of respiration. Br. J. Anaesth. 2008, 100, 747–758.
- Manzke, T.; Niebert, M.; Koch, U.; Caley, A.; Vogelgesang, S.; Bischoff, A.-M.; Hülsmann, S.; Ponimaskin, E.; Müller, U.; Smart, T.; et al. Die von serotoninrezeptor 1A modulierte dephosphorylierung des glyzinrezeptors α3. Der. Schmerz 2011, 25, 272–281.
- Dunn, E.B.; Wolfe, J.J. Finding an optimal dose: Considerations in accurate opioid dispensing. Vet. Hum. Toxicol. 2000, 42, 36–38.
- Lewanowitsch, T.; Irvine, R.J. Naloxone methiodide reverses opioid-induced respiratory depression and analgesia without withdrawal. Eur. J. Pharmacol. 2002, 445, 61–67.
- Droney, J.M.; Gretton, S.K.; Sato, H.; Ross, J.R.; Branford, R.; Welsh, K.I.; Cookson, W.; Riley, J. Analgesia and central side-effects: Two separate dimensions of morphine response. Br. J. Clin. Pharmacol. 2013, 75, 1340–1350.
- Babbini, M.; Davis, W.M. Time-dose relationships for locomotor activity effects of morphine after acute or repeated treatment. Br. J. Pharmacol. 1972, 46, 213–224.
- Hollais, A.W.; Patti, C.L.; Zanin, K.A.; Fukushiro, D.F.; Berro, L.F.; Carvalho, R.C.; Kameda, S.R.; Frussa-Filho, R. Effects of acute and long-term typical or atypical neuroleptics on morphine-induced behavioural effects in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 255–263.
- Patti, C.L.; Frussa-Filho, R.; Silva, R.; Carvalho, R.C.; Kameda, S.R.; Takatsu-Coleman, A.L.; Cunha, J.L.; Abílio, V.C. Behavioral characterization of morphine effects on motor activity in mice. Pharmacol. Biochem. Behav. 2005, 81, 923–927.
- Rodrı́guez-Arias, M.; Broseta, I.; Aguilar, M.; Miñarro, J. Lack of specific effects of selective D1 and D2 dopamine antagonists vs. risperidone on morphine-induced hyperactivity. Pharmacol. Biochem. Behav. 2000, 66, 189–197.
- Domino, E.F.; Vasco, M.R.; Wilson, A.E. Mixed depressant and stimulant actions of morphine and their relationship to brain acetylcholine. Life Sci. 1976, 18, 361–376.
- Brady, L.S.; Holtzman, S.G. Locomotor activity in morphine-dependent and post-dependent rats. Pharmacol. Biochem. Behav. 1981, 14, 361–370.
- Tulunay, F.C.; Ayhan, I.H.; Sparber, S.B. The effects of morphine and delta-9-tetrahydrocannabinol on motor activity in rats. Psychopharmacology 1982, 78, 358–360.
- Murphy, N.P.; Lam, H.A.; Maidment, N.T. A comparison of morphine-induced locomotor activity and mesolimbic dopamine release in C57BL6, 129Sv and DBA2 mice. J. Neurochem. 2008, 79, 626–635.
- Christie, J.E.; Crow, T.J. Turning behaviour as an index of the action of amphetamines and ephedrines on central dopamine-containing neurones. Br. J. Pharmacol. 1971, 43, 658–667.
- Barber, D.; Blackburn, T.; Greenwood, D. An automatic apparatus for recording rotational behaviour in rats with brain lesions. Physiol. Behav. 1973, 11, 117–120.
- Urs, N.M.; Daigle, T.L.; Caron, M.G. A Dopamine D1 receptor-dependent β-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology 2010, 36, 551–558.
- Ryczko, D.; Dubuc, R. Dopamine and the brainstem locomotor networks: From lamprey to human. Front. Neurosci. 2017, 11, 295.
- Chastain, L.G.; Qu, H.; Bourke, C.H.; Iuvone, P.M.; Dobner, P.R.; Nemeroff, C.B.; Kinkead, B. Striatal dopamine receptor plasticity in neurotensin deficient mice. Behav. Brain Res. 2014, 280, 160–171.
- Borgkvist, A.; Usiello, A.; Greengard, P.; Fisone, G. Activation of the cAMP/PKA/DARPP-32 signaling pathway is required for morphine psychomotor stimulation but not for morphine reward. Neuropsychopharmacology 2007, 32, 1995–2003.
- Mishra, A.; Singh, S.; Shukla, S. Physiological and functional basis of dopamine receptors and their role in neurogenesis: Possible implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12, 1179069518779829.
- Horsfall, J.T.; Sprague, J.E. The pharmacology and toxicology of the ‘Holy Trinity’. Basic Clin. Pharmacol. Toxicol. 2016, 120, 115–119.
- Anderson, E.; Hearing, M. Chapter 4-Neural circuit plasticity in addiction. In Neural Mechanisms of Addiction; Torregrossa, M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 35–60.
- Oliva, I.; Wanat, M.J. Ventral tegmental area afferents and drug-dependent behaviors. Front. Psychiatry 2016, 7, 30.
- Adinoff, B. Neurobiologic processes in drug reward and addiction. Harv. Rev. Psychiatry 2004, 12, 305–320.
- Gardner, E.L.; Ashby, C.R. Heterogeneity of the mesotelencephalic dopamine fibers: Physiology and pharmacology. Neurosci. Biobehav. Rev. 2000, 24, 115–118.
- Lever, C.; Burton, S.; O’Keefe, J. Rearing on hind legs, environmental novelty, and the hippocampal formation. Rev. Neurosci. 2006, 17, 111–133.
- Rodriguiz, R.M.; Wetsel, W.C. Frontiers in neuroscience assessments of cognitive deficits in mutant mice. In Animal Models of Cognitive Impairment; Levin, E.D., Buccafusco, J.J., Eds.; CRC Press/Taylor & Francis, Taylor & Francis Group, LLC.: Boca Raton, FL, USA, 2006.
- Alves, R.; de Carvalho, J.G.B.; Venditti, M.A.C. High-and Low-Rearing rats differ in the brain excitability controlled by the allosteric benzodiazepine site in the GABAA receptor. J. Behav. Brain Sci. 2012, 2, 315–325.
- Valverde, O.; Mantamadiotis, T.; Torrecilla, M.; Ugedo, L.; Pineda, J.; Bleckmann, S.; Gass, P.; Kretz, O.; Mitchell, J.M.; Schütz, G.; et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology 2004, 29, 1122–1133.
- Contet, C.; Filliol, D.; Matifas, A.; Kieffer, B.L. Morphine-induced analgesic tolerance, locomotor sensitization and physical dependence do not require modification of mu opioid receptor, cdk5 and adenylate cyclase activity. Neuropsychopharmacology 2008, 54, 475–486.
- Zan, G.-Y.; Wang, Q.; Wang, Y.-J.; Liu, Y.; Hang, A.; Shu, X.-H.; Liu, J.-G. Antagonism of κ opioid receptor in the nucleus accumbens prevents the depressive-like behaviors following prolonged morphine abstinence. Behav. Brain Res. 2015, 291, 334–341.
- Aceves, M.; Bancroft, E.; Aceves, A.R.; Hook, M.A. Nor-binaltorphimine blocks the adverse effects of morphine after spinal cord injury. J. Neurotrauma 2017, 34, 1164–1174.
- Walsh, R.N.; Cummins, R.A. The open-field test: A critical review. Psychol. Bull. 1976, 83, 482–504.
- Paul, A.K.; Gueven, N.; Dietis, N. Age-dependent antinociception and behavioral inhibition by morphine. Pharmacol. Biochem. Behav. 2018, 168, 8–16.
- Paul, A.K.; Gueven, N.; Dietis, N. Data on prolonged morphine-induced antinociception and behavioral inhibition in older rats. Data Brief. 2018, 19, 183–188.
More