MERTK and AXL are members of the TAM family of receptor tyrosine kinases and are abnormally expressed in 69% and 93% of non-small cell lung cancers (NSCLCs), respectively. Expression of MERTK and/or AXL provides a survival advantage for NSCLC cells and correlates with lymph node metastasis, drug resistance, and disease progression in patients with NSCLC. The TAM receptors on host tumor infiltrating cells also play important roles in the immunosuppressive tumor microenvironment. Thus, MERTK and AXL are attractive biologic targets for NSCLC treatment.
- MERTK
- AXL
- TAM family
- NSCLC
1. Introduction
Lung cancers are divided into two broad categories: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Following clinical validation of translational inhibitors targeting two important NSCLC oncogenic drivers, epidermal growth factor receptor (EGFR) [1][2][3][4][15,16,17,18] and anaplastic lymphoma kinase (ALK) [5][6][7][19,20,21], molecular-targeted therapies have been applied to the management of metastatic NSCLC, resulting in remarkably improved prognosis and quality of life relative to patients treated with conventional chemotherapeutics [8][9][10][22,23,24]. Additional mutated oncogenic proteins have been identified in NSCLC, including HER2, BRAF, RET, MET, and ROS1 [6][11][20,25]. Even though patients respond to targeted therapies initially, the majority of patients, if not ultimately all patients, relapse within 1 to 2 years when treated with targeted therapies [12][13][14][15][16][26,27,28,29,30]. Therefore, understanding the mechanisms of primary and secondary resistance to current targeted therapies is critical to enhance patient outcomes. New therapeutic approaches will be required to further enhance outcomes. Both MERTK and AXL, members of the TAM (TYRO3, AXL, and MERTK) family of receptor tyrosine kinases (RTK), are emerging therapeutic targets in NSCLC.
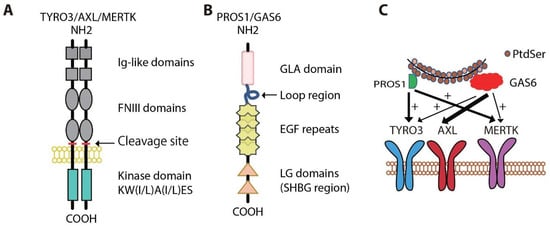
2. Physiologic Roles for MERTK and AXL
3. Oncogenic Roles for MERTK and AXL
3.1. Roles in NSCLC
MERTK and AXL are frequently aberrantly expressed in NSCLC patient samples, but are absent or expressed at low levels in normal human bronchial epithelial cells [37][38][39][40][41][42][43][44][51,52,53,54,55,56,57,58]. High levels of AXL have been described in subsets of both treatment-naïve and relapsed NSCLC [44][45][46][58,59,60]. Increased AXL expression was associated with increased tumor cell invasiveness and tumor grade and predicted poorer survival in patients with NSCLC [42][43][47][48][49][50][51][56,57,61,62,63,64,65]. Inhibition of MERTK in NSCLC cell lines with a small molecule MERTK tyrosine kinase inhibitor (TKI), MERTK-specific blocking monoclonal antibody, or shRNA induced apoptosis and decreased colony formation in vitro and inhibited tumor growth in vivo [52][53][54][55][66,67,68,69]. Treatment with an antibody against active AXL or siRNA/shRNA AXL knockdown also provided anti-tumor activity in NSCLC models [42][50][56][56,64,70].
High levels of MERTK and/or AXL have also been implicated in drug resistance and radioresistance [40][41][46][55][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][54,55,60,69,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86]. Increased MERTK or AXL expression in NSCLC correlated with chemotherapy resistance [37][52][56][73][74][75][51,66,70,87,88,89]. Conversely, MERTK or AXL knockdown, treatment with a MERTK monoclonal antibody, or AXL inhibitor R428 or MP-470 promoted apoptosis and increased the sensitivity of NSCLC cells to chemotherapeutic agents [37][52][56][73][74][75][51,66,70,87,88,89]. Upregulated AXL and its interaction with EGFR were associated with resistance to PI3Kα inhibition due to sustained mTOR activation, and addition of AXL inhibitor R428 sensitized tumor cells to PI3Kα [62][76]. In addition, AXL has been implicated in resistance to anti-IGF-1R therapy [76][77][90,91] and resistance to BRAF/MEK inhibitors [78][92].
3.2. Functions in Cancer Cells
3.3. Signaling in Cancer Cells
Activation of AXL and/or MERTK leads to signaling cascades that are important for tumor progression (Figure 2). MERTK kinase activity is associated with phosphorylation at three tyrosine residues: Y749, Y753, and Y754 on MERTK [98][131] and Y779, Y821, and Y866 on AXL [99][100][132,133]. Both Y779 and Y821 on AXL and two additional phosphorylation sites for MERTK (Y872 and Y929) are docking sites for GRB2 and the p85 regulatory subunit of PI3K, which activate MEK/ERK and PI3K/AKT signaling pathways, respectively [82][99][100][109,132,133]. The MEK/ERK signaling is associated with cell proliferation [101][134], while the PI3K/AKT pathway is preferentially involved in tumor cell survival [102][135]. MERTK-dependent cell migration is mediated by FAK signaling [41][103][55,136], while MERTK induced transformation correlates with activation of STAT-dependent transcription [104][137]. The anti-apoptotic effects of MERTK also correlate with negative regulation of the pro-apoptotic tumor suppressor WW domain-containing oxidoreductase (Wwox) [105][138]. Also, AXL inhibition is reported to mediate apoptosis by reducing the expression of the anti-apoptotic protein MCL1 [106][139]. AXL dimerizes with and phosphorylates EGFR to promote activation of the PLCγ-PKC-mTOR signaling cascade and tumor cell survival [62][76]. Similarly, there is crosstalk between MERTK and EGFR and they are frequently co-expressed on both mtEGFR- and wtEGFR-expressing NSCLC cell lines [55][66][69,80]. In fact, MERTK stabilized the EGFR protein on the cell surface, probably by preventing EGFR internalization and degradation, as EGF-dependent EGFR turnover was reversed by inhibition of lysosomal hydrolase activity [107][140]. Further, inhibition of MERTK expression using siRNA destabilized expression of EGFR protein.3.4. Immune Regulatory Functions in the Tumor Microenvironment

3.4. Immune Regulatory Functions in the Tumor Microenvironment
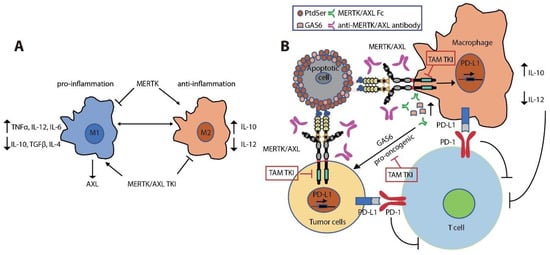
MERTK is upregulated upon monocyte to macrophage differentiation [108][109][110][141,142,143]. Expression of MERTK and AXL on tumor-infiltrating macrophages polarizes them towards a pro-tumor M2-like phenotype [110][111][112][113][143,144,145,146]. M2 macrophages promote an immunosuppressive tumor microenvironment by increasing expression of wound-healing cytokines (IL-10, TGFβ, and IL-4) and decreasing pro-inflammatory cytokines (IL-12, TNFα, and IL-6) [31][110][114][115][116][117][118][45,143,147,148,149,150,151] (Figure 3A). MERTK activation negatively regulates the secretion of pro-inflammatory cytokines, such as TNFα, through suppression of NFκB activation in macrophages [31][119][45,152]. LPS challenge led to over-produced TNFα in Mertkkd mice, which lack the tyrosine kinase signaling domain, due to hyper-activation of NFκB [31][120][45,153]. Inhibition of MERTK by knockout of Mertk in mice, neutralization of TAM kinase signaling using a recombinant MERTK-Fc protein as a ligand sink or a GAS6 blocking antibody, and knockout of AXL in macrophages also impaired M2-macrophage anti-inflammatory phenotypes, decreased immunosuppressive IL-10 production, and increased pro-inflammatory IL-12 release [72][110][121][122][86,143,154,155]. These cytokine alterations lead to expansion of anti-tumor CD8+ T lymphocytes and inhibition of tumor growth and metastasis (Figure 3B). Indeed, inhibition of MERTK in the tumor microenvironment in Mertk−/− mice was sufficient to decrease tumor growth and metastasis [121][154]. MERTK-expressing dendritic cells can also regulate T cell activation directly [123][156]. Blocking MERTK on dendritic cells using anti-MERTK antibody promoted T cell proliferation, while treatment with a MERTK-Fc protein to mimic the effect of MERTK expressed on human dendritic cells suppressed naïve CD4+ T cell proliferation [123][156]. The anti-inflammatory effect of MERTK activation in macrophages and apoptotic cell-treated dendritic cells was mediated by inhibition of NFκB activation [36][124][125][50,157,158] or by induction of toll-like receptor (TLR) suppressor of cytokine signaling 1 (SOCS1) and SOCS3 [125][126][127][158,159,160] (Figure 2). Further, the MERTK ligand PROS1 also promotes resolution of inflammation by macrophages and inhibits macrophage M1 polarization to reduce anti-tumor immune response [128][161].