Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Lynn M. Pezzanite and Version 2 by Catherine Yang.
The innate immune system is classically triggered by host responses to pathogen-associated molecular patterns (PAMPs) induced by interactions with invariable pattern-recognition receptors (PRRs) on synovial joint immune cells such as neutrophils, macrophages, monocytes, and dendritic cells (DC). PRRs are comprised of a family of cell surface, endosomal and cytosolic receptors, including Toll-like receptors and NOD-like receptors. Activation of PRRs within tissues such as the joint leads initially to rapid-onset inflammatory responses, followed later by initiation of adaptive immune responses and finally by healing responses in the case of tissue injuries.
- osteoarthritis
- innate immunity
1. Pattern Recognition Receptors and Immune Cells in Joints
Although understanding of OA pathogenesis is still evolving, innate immune cells, particularly myeloid cells, play a role in regulating and perpetuating low-grade inflammation that characterizes Osteoarthritis (OA) (Figure 1). Histologically, synovial inflammation in OA is characterized by transient and/or cyclical hyperplasia of the synovial lining cells accompanied by inflammatory cell infiltration. This population consists predominantly of macrophages and smaller numbers of T and B cells, mast cells, and natural killer (NK) cells [1][2][3][4][15,16,17,18].
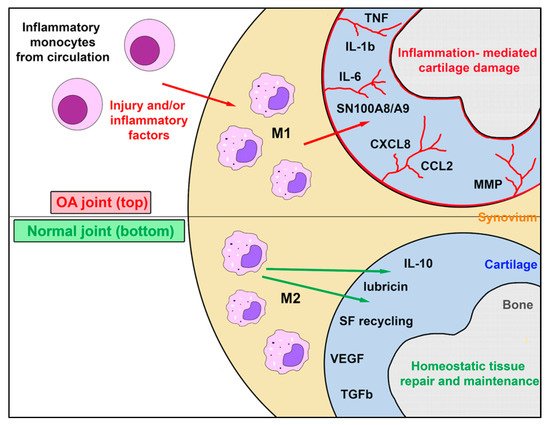
Figure 1. Role of synovial resident macrophages and inflammatory macrophages in joint health and osteoarthritis. This figure illustrates the complex interaction between endogenous self-renewing synovial cell macrophages, which exist as M2 cells in the healthy joint, and the inflammatory macrophage M1 population recruited from inflammatory monocytes in blood in response to chemokines produced during joint inflammation occurring during the progression of OA. While M1 macrophages may assume an M2 phenotype as inflammation subsides, endogenous M2 macrophages typically do not generate M1 macrophages. The factors secreted by synovial M1 and M2 macrophage populations in health and disease are complex and largely distinct. Abbreviations: tumor necrosis factor (TNF); interleukin 1Β (IL-1Β); interleukin 6 (IL-6), Myeloid protein SN100A8/A9; C-X-C Motif Chemokine Ligand 8 or Interleukin-8 (CXCL8); (CCL2); matrix metalloproteinase (MMP); interleukin 10 (IL-10); synovial fluid (SF); vascular endothelial growth factor (VEGF); transforming growth factor B (TGF-B).
The innate immune system is classically triggered by host responses to pathogen-associated molecular patterns (PAMPs) induced by interactions with invariable pattern-recognition receptors (PRRs) on synovial joint immune cells such as neutrophils, macrophages, monocytes, and dendritic cells (DC). PRRs are comprised of a family of cell surface, endosomal and cytosolic receptors, including Toll-like receptors and NOD-like receptors [5][19]. Activation of PRRs within tissues such as the joint leads initially to rapid-onset inflammatory responses, followed later by initiation of adaptive immune responses and finally by healing responses in the case of tissue injuries.
In addition to invading pathogens, PRRs also recognize various endogenous “danger signals,” known as damage associated molecular patterns (DAMPs), triggered from cell or tissue damage. Non-infectious DAMPs (e.g., cartilage degradation products) can activate macrophages and DCs, including inflammasome activation, initiation of cellular pyroptosis, and other unfolded protein responses that induce inflammatory, metabolic, and adaptive immune pathologies [6][7][20,21]. The list of known DAMPs is rapidly growing and beyond the scope of this review. Studies have shown that cartilage injury results in the release of DAMPs within the joint, including breakdown products from fibronectin and hyaluronan [8][9][10][22,23,24].
2. Macrophages and OA
Macrophages are central players in host defense and are distributed in almost all tissues, having unique functions in each organ depending on the specific microenvironment that influences their functional properties. In addition to their more familiar functions as proinflammatory, scavenger, antimicrobial, and antitumor defense effector/mediators, macrophages also function as immune modulators, promoting anti-inflammatory and tissue repair processes [11][12][13][14][15][25,26,27,28,29]. Given their central role in OA, a clinical approach targeting activated macrophages at an earlier stage of OA may serve to inhibit or slow the progression of disease [16][17][30,31].
Tissue macrophages such as alveolar macrophages and Kupffer cells are derived from the yolk sac during early embryogenesis and serve as a self-renewing population of tissue macrophages throughout life. It is likely that synovial macrophages are also a unique tissue specific population of self-renewing cells, which have an important role in maintaining articular homeostasis. Inflammatory monocytes by contrast reach the joint following release from the bone marrow and differentiate into short-lived inflammatory macrophages in synovial tissues (Figure 1). Macrophages in tissues are classified as proinflammatory (M1) or anti-inflammatory (M2) cells [18][32]. In rheumatoid arthritis, the majority of M1 macrophages are bone marrow-derived and are recruited to and differentiate within the joint. In vivo heterogeneity varies between M1 and M2 populations have been identified using next-generation sequencing technologies and transcriptomic analysis [19][9]. The rationale for having seemingly opposing systems is to achieve immune homeostasis via a proper balance of M1 and M2 responses, thereby maintaining a tightly controlled immune environment capable of optimal protection while preventing host tissue destruction. This is a very important consideration as macrophages are the more numerous immune cells in the synovium and further understanding is paramount before they can be exploited for therapeutic purposes [19][11][20][9,25,33].
M1 macrophages typically have multiple functions and are responsible for the release of molecules that can drive inflammation. M1 macrophages are differentially stimulated by NK cells and NK T cell secretion of IFNγ, by various TLR agonists such as bacterial lipopolysaccharide (LPS), or growth factors such as granulocyte-macrophage-colony-stimulating factor (GM-CSF). M2 macrophages, on the other hand, are functionally considered to be more anti-inflammatory and viewed as mediating an opposing immune regulatory response that counters proinflammatory M1 responses and abrogates host tissue destruction. M2 macrophages are stimulated by IL-4 and IL-13 secreted primarily by Th2 T cells, mast cells and, more recently discovered, by basophils—to produce mostly anti-inflammatory cytokines such as IL-10, IL-1ra, TGFβ, and arginase-1 (Arg-1) [13][21][27,34]. Under a homeostatic state, most macrophages display the M2 phenotype to maintain tissue surveillance and protect against metabolically derived oxidative conditions and inflammation [22][35]. Specifically, CD206+ M2 macrophages produce IL-4, IL-10, and MMP-12, which reduces neutrophil influx to the joint and can counter the inflammatory response and catabolic effects seen in OA [19][23][11][12][14][15][24][25][26][27][28][29][30][9,10,25,26,28,29,36,37,38,39,40,41,42].
When activated, CD80/86+ M1 macrophages secrete high levels of proinflammatory cytokines such as IL-1β, IL-6, IL-8, IL-12, TNFα, and alarmins; they also induce Th1 adaptive immune responses [11][25]. These synovial macrophages, in turn, activate the production of harmful molecules such as matrix metalloproteinases (MMP-1, MMP-3, MMP-8, MMP-9, MMP-13) from other innate immune system cells and synovial fibroblasts, causing generation of DAMPs from extra cellular matrix (ECM) degeneration [11][25]. Additionally, pro-inflammatory cytokines stimulate synthesis of PGE2 by cyclo-oxygenase-2, microsomal PGE synthase-1, and soluble phospholipase A2 [31][11][1,25] This is associated with production of nitric oxide (NO) by nitric oxide synthetase, reactive oxygen species (ROS), 5-lipoxygenase and leukotriene B4 [11][25]. This pro-inflammatory environment also favors the activation of neuropeptides, such as substance P, that increase inflammation and result in joint pain [31][11][1,25].
Over-production of M1-derived cytokines, growth factors, various proteases, and oxidizers in inflamed synovium can heavily contribute to the initiation and progression of OA via DAMP-driven cellular pyroptosis [11][32][33][34][25,43,44,45]. DAMP activation of synovial macrophages in OA produces ROS that may incite the Nod-like receptor protein 3 (NLRP3) component of the caspase-1-activating inflammasome, mediating secretion of pro-inflammatory cytokines IL-1β, IL-18, and TNFα [11][25]. DAMPs identified in this process include degradation products from hyaluronidase, hydroxyapatite (HA) and basic calcium phosphate (BCP) crystals from cartilaginous calcification [11][25]. Although inflammasomes in veterinary species have not been well characterized, recent work has demonstrated that equine peripheral blood mononuclear cells (PBMCs) normally secrete IL-1β in response to well-known inflammasome activators of NLRP3 [32][33][34][43,44,45]. The key transcription factor countering the resulting oxidative stress is nuclear factor E2-related factor 2 (Nrf2), which protects against oxidative stress and tissue damage. Accordingly, future research into the signaling mechanisms transducing the Nrf2-mediated transcriptional program may serve to develop novel therapies for OA [35][36][37][38][39][46,47,48,49,50].
It should be noted that, in vivo, macrophages do not adhere to strict dichotomous phenotypes, but rather express plasticity across the spectrum between M1 and M2, capable of signaling for either inflammation or healing depending on tissue milieu [11][25]. For example, availability of various DAMPs within the joint microenvironment, their anatomical origin, and the tissue in which they are located, may influence macrophage phenotype, making it difficult to ensure that all CD86+ macrophages exclusively perform M1 functions and that all CD206+ macrophages exclusively assist in healing and resolution of inflammation. Thus, caution should be taken when describing the overall pro- vs. anti-inflammatory status within a locale [19][12][9,26]. Furthermore, studies involving murine models of macrophage depletion have reported mixed results, suggesting that this therapy might vary greatly between tissues and might not always be beneficial as all populations of myeloid cells are affected, including DCs and neutrophils [19][9].
3. Value of Equine Models for OA Research
Many factors can influence the outcome of data acquired from the various animal models of OA, such as overall intrinsic species and strain variation, age, sex, housing, time of intervention, stress levels, and activity. These are important considerations that must be taken into account when assessing the outcome of any project involving knowledge gathered regarding the predisposition, cause, and ultimate therapeutic success of a given research approach [40][51]. In vitro models are affordable and make it easier to control many variables at the same time, but the bi-dimensional model does not fully represent what truly happens in the joint. Three-dimensional models, including explants, scaffold-based and scaffold-free systems, provide a more similar environment to the joint and its interaction with the cells while allowing more variable control. In vivo models, especially large animals, remain a more viable option due to anatomic similarities and naturally occurring OA [41][52].
Although data from murine models is very informative and valuable in biomedical research, the variance in the programmed genomic responses to acute inflammatory responses and their significant anatomical differences might not make them the best translational option for human or equine patients [42][53]. From an anatomical perspective, the horse model of OA most closely resembles humans with regard to articular cartilage thickness [43][54]. In addition, horses suffer from OA frequently and spontaneously, providing veterinarians with significant experience in treating the condition. The equine carpal chip model of OA represents a predictable model with which to test novel approaches such as immunomodulating therapies like mesenchymal stromal cells (MSCs) [44][55]. Accordingly, horses are one of the best animals to use as a model for human OA, due to similarities in many of the joints’ movement and comparable cartilage and subchondral bone thicknesses [45][46][56,57].
4. Equine Innate Immune System and OA
Of the structures present in the joint, the most relevant regarding its capacity to mount an inflammatory response is the synovium. The cells that are recruited and react to post-traumatic osteoarthritis are components of the innate immune system and are mainly resident macrophages. Destruction of cartilage matrix by MMPs will result in the immediate production of IL-1β and TNFα, and these powerful pro-inflammatory cytokines can perpetuate the inflammatory cascade and pain in the joint [25][47][37,58]. Although there is a strong correlation between obesity and chronic inflammation in humans and mice [48][59], such a comparison has not been made for horses and it is likely that post-traumatic injury is the more common cause of OA in horses.
Synovium and innate immunity—The synovium is the major site of articular inflammation in OA and is often marked by hyperplasia of the synovial lining cells coupled with infiltration of inflammatory cells consisting mostly of macrophages and smaller numbers of other cells [1][2][3][4][15,16,17,18]. The macrophage population in the synovium is classified into two groups: 25% of the cells are type A synovial cells (macrophages) and 75% of the cells are type B synoviocytes (fibroblast-like) [49][12]. Also, macrophages can be found in synovial fluid and can account for 70% of the total cells in a non-inflamed joint, with these numbers increasing up to 90% in an inflammation model for repeated arthrocentesis [50][13]. Thus, immune cells constitute a significant percentage of all the cells in joint tissues and synovial fluid.
Cartilage and innate immunity—Mature equine cartilage predominantly contains chondrocytes, including only a very small population of progenitor cells. Cartilage is a firm yet smooth, lubricated, and almost frictionless surface that enables normal joint function and depends on the synovium for lubrication. It is important to consider that cartilage is an aneural and avascular tissue, depending directly for their metabolic support on factors present in synovial fluid. Cartilage also reacts to inflammatory reactions triggered in the synovium. It is also important to note that cartilage degradation products in synovial fluid, as well as micro-fissures in articular tissue, are often present before any degeneration can be detected using current imaging technology. It has been speculated that early cartilage degradation events may drive the development of inflammation within OA synovium, which happens through the activation of resident macrophages through DAMPS favoring the release of TNFα and IL-1β that will, in turn, increase the MMPs production by the chondrocytes [23][50][10,13].
Subchondral bone and innate immunity—Subchondral bone differs significantly from cartilage, as it is heavily vascularized, allowing for major tissue turnover and the ability to remodel to adapt to mechanical loads. Inflammation of the subchondral bone can lead to the production of angiogenic factors and local MMP production, which is thought to stimulate cartilage degeneration and the formation of osteophytes [23][51][52][10,60,61].