Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Stefano Carugo.
Arrhythmogenic cardiomyopathy (ACM) is an inherited heart disease characterized by sudden death in young people and featured by fibro-adipose myocardium replacement, malignant arrhythmias, and heart failure. The Ca2+ toolkit is heavily remodeled in cardiomyocytes derived from a mouse model of ACM defective of the desmosomal protein plakophilin-2. Furthermore, ACM-related mutations were found in genes encoding for proteins involved in excitation‒contraction coupling, e.g., type 2 ryanodine receptor and phospholamban.
- arrhythmogenic cardiomyopathy
- Ca2+
1. Introduction
The heart is a highly specialized machine that requires fine regulation: intracellular changes and cell-to-cell communication disturbances could destroy this balance. Depending on the type of cardiac arrhythmia, a series of events may occur: intracellular dysfunction‒tissue remodeling‒impaired cell‒cell connection–electrical dysfunction. Loss of cardiac tissue integrity could be both a cause and an effect of arrhythmic phenotype. A typical example is arrhythmogenic cardiomyopathy (ACM), a cardiac disorder in which mutations in proteins of the desmosome may be present, causing cell-to-cell discontinuity, arrhythmic events, and myocardial fibro-adipose substitution [1,2,3][1][2][3].
Intracellular calcium (Ca2+) signals drive excitation‒contraction (EC) coupling and are, therefore, necessary for the heart to effectively pump blood into the pulmonary artery and aorta. It is, therefore, not surprising that any defect in the Ca2+ cycling machinery or in the complex network of Ca2+-related proteins that maintain cardiac Ca2+ homeostasis under tight control may severely compromise cardiac function. Alterations or mutations in the Ca2+ handling machinery have been associated with a number of severe cardiac diseases, including cardiac hypertrophy, dilated cardiomyopathy, and heart failure [4[4][5],5], and several inherited arrhythmia syndromes (i.e., Timothy syndrome, Brugada syndrome, and early repolarization syndromes) [6,7,8][6][7][8].
2. Calcium Signaling and Cardiac Arrhythmias: A Tight Connection
Cardiac contraction is triggered by a transient increase in intracellular Ca2+ concentration ([Ca2+]i) induced by membrane depolarization according to a process known as EC coupling [6,7,9][6][7][9]. Briefly, as the action potential, triggered by an influx of sodium (Na+) via the voltage-gated Na+ channels, propagates along the T-tubules, membrane depolarization opens CaV1.2, which is the pore-forming α-subunit of L-type voltage-gated Ca2+ channels (VGCCs), thereby causing the influx of Ca2+ into the dyadic junction, which separates the sarcolemma from the closely apposed (~15 nm) junctional sarcoplasmic reticulum (SR) [10] in ventricular myocytes [9]. Extracellular Ca2+ influx causes a large increase in local [Ca2+]i, which activates a cluster (7–20) of type 2 Ryanodine receptors (RYR2) in a process known as Ca2+-induced Ca2+ release (CICR) and gives rise to significant Ca2+ release events from the junctional SR (called Ca2+ sparks). The temporal summation of these local events by the propagating action potential results in a regenerative Ca2+ transient that is detected by troponin C to initiate the sliding of the thick (myosin) and thin (actin) filaments, cell shortening, and hence pressure development within each ventricle and ejection of blood into the circulation during cardiac systole [6,7,9][6][7][9]. Subsequently, [Ca2+]i drops and mechanical force relaxes quickly to pre-systolic levels during cardiac diastole, which is essential to enable cardiac chambers to refill with blood. The increase in [Ca2+]i is indeed short-lived as cytosolic Ca2+ is extruded across the plasma membrane by the Na+/Ca2+ exchanger (NCX1) and the plasmalemmal Ca2+-ATPase (PMCA) and sequestered back into the SR by the sarco-endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) [6,7,9][6][7][9]. The Ca2+ affinity of SERCA2a is regulated by the phosphoprotein phospholamban (PLN), which increases or reduces the pumping rate of SERCA2a depending on its phosphorylation state. In the dephosphorylated state, PLN binds to SERCA2a and inhibits the Ca2+ transport ability of the pump by reducing its apparent affinity for Ca2+. However, when PLN is phosphorylated, it dissociates from SERCA2a, thereby increasing the pump’s affinity for Ca2+ and favoring relaxation [11]. PLN presents two sites of phosphorylation: Ser16 for protein kinase A (PKA) and Thr17 for Ca2+/Calmodulin-dependent protein kinase II (CaMKII) and protein kinase B (AKT) [12]. Importantly, β-adrenergic stimulation exerts a profound impact on cardiac Ca2+ handling by modulating CaV1.2, RYR2 and SERCA2a activity. Accordingly, stimulation of β-adrenergic receptors, e.g., with isoproterenol, engages PKA and CaMKII. PKA, in turn, phosphorylates CaV1.2 and PLN, thereby increasing the amplitude of L-type voltage-gated Ca2+ currents and boosting SR Ca2+ uptake. Furthermore, Bovo and colleagues recently demonstrated that PKA-dependent phosphorylation also regulates RYR2-mediated SR Ca2+ release independently of PKA effects on SR Ca2+ load [13].3. Arrhythmogenic Cardiomyopathy as Adhesion Disorder: Ca2+-Dependent Desmosomes’ Stability
Cell to cell junctions are essential to confer stability to tissues, especially those undergoing continuous mechanical stretch, such as the heart and skin. The myocardium tissue integrity is based on desmosomes, anchoring cell junctions that are assembled in strong and highly specialized complexes. Loss or mutations of cell junctions are associated with human genetic diseases and ACM [39,53,78,79][14][15][16][17]. Desmosomes consist of specific cadherins, i.e., DSC2 and desmoglein-2 (DSG2), which act like a bridge to join the lateral edges of neighboring cells. The desmosomal cadherins bind proteins of the armadillo family, PG and PKP2, that are anchored to desmoplakin (DSP), the main intracellular component responsible for the adhesion to the intermediate filament network [80][18]. Desmosomes undergo a transition from hyper- to low-adhesion states depending on extracellular Ca2+ levels. The Ca2+-independent adhesive state of desmosomes is referred to as hyper-adhesion-state since it represents a higher-affinity and more stable binding ability both for desmosomes and adherens junctions. During tissue remodeling, wounding, and cell migration, a lower-affinity adhesive state is required and desmosomes adopt the Ca2+-dependent state, losing their organized structure [81,82,83][19][20][21] (Figure 21). This transition does not involve protein composition rearrangement, but it can influence the cadherin packing. Based on the nature of molecules underlying the desmosomal adhesion, both the formation and disruption are Ca2+-dependent mechanisms [84,85,86][22][23][24]. The desmosomal cadherins, DSC2 and DSG2, and the classical cadherin, E-cadherin, represent the Ca2+-dependent components of desmosomes and adherens junctions, respectively [87][25]. In vitro, the Ca2+-dependent mechanism is linked to the culture confluence state: in a confluent cell monolayer, desmosomes are Ca2+-independent until the confluence is destroyed by wounding the cell sheet, thereby resulting in the propagation of Ca2+ dependence through the whole monolayer [81][19].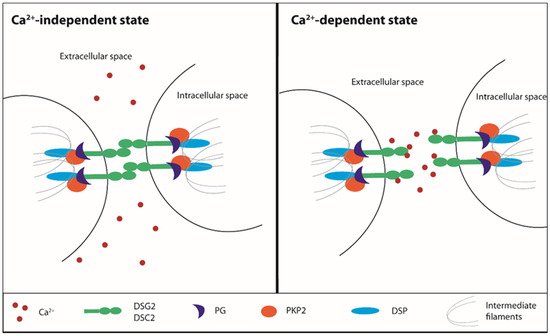
Figure 21. Ca2+-dependent desmosomes stability. Ca2+: calcium, DSG2: desmoglein 2, DSC2: desmocollin 2, PG: plakoglobin, PKP2: plakophilin 2, DSP: desmoplakin.
4. Desmosomal Mutations and Arrhythmogenic Cardiomyopathy: Derangement of the Ca2+ Toolkit as a Consequence Rather than a Pathogenic Defect
The mechanism that induces the reduction of adhesion during tissue remodeling is not completely clear, but desmosomes can be totally internalized by cells. After the internalization, they are degraded or disassembled in order to recycle their component proteins [88,89][26][27]. During the Ca2+-dependence state, the desmosomes undergo a conformational rearrangement of the cadherin extracellular domains that induces a less organized structure and a more easily adhesive binding disruption [90][28]. The molecular basis of the adhesion between desmosomes involves the activation of protein kinase C (PKC) or inhibition of protein phosphatases [82,83][20][21]. The treatment with a low-Ca2+ medium induces loss of intercellular adhesion and the formation of half-desmosomes: the broken desmosomes are internalized by a PKC-dependent mechanism and transported to the centrosome, where they undergo proteasomal and lysosomal degradation [89][27]. The central role of PKCα is supported by experiments performed on a mouse model lacking or overexpressing this kinase. Both in absence and following inhibition of PKCα, the switch to Ca2+-dependence is blocked and hyperadhesive desmosomes lock the cells together, preventing cell motility and epithelial migration. On the contrary, in mice overexpressing a constitutively active PKCα, a rapid transition to a Ca2+ dependence state make desmosomes weakly adhesive, thus facilitating cell mobilization and promoting re-epithelialization [91][29].
Desmosomal mutations have been identified as a crucial determinant of ACM since the late 1980s [92][30]. A homozygous truncating mutation in the JUP gene, encoding for PG, was first identified through genetic linkage analysis as a typical syndromic form of right-sided ACM, known as Naxos disease [93][31]. Subsequently, genetic analysis revealed that another desmosomal gene, DSP, encoding for desmoplakin, harbored a homozygous truncating mutation in ACM in patients with a cardiocutaneous syndrome, named Carvajal syndrome [93,94][31][32]. These earlier discoveries sparked the search for causative mutations in additional desmosome genes in ACM patients and led to the identification of either missense or truncating mutations in the following desmosome genes: PKP2 (encoding for plakophilin 2), DSC2 (encoding desmocollin 2), and DSG2 (encoding for desmoglein 2) (Table 1) [93][31]. It has been estimated that mutations in desmosomal genes are present in approximately half of ACM patients, with PKP2 being the most commonly affected gene in adults [93][31]. The molecular mechanisms whereby PKP2 mutations cause ACM are yet to be fully clarified, but no change in the Ca2+-sensitivity of the desmosomal junctions has been reported [93,95][31][33]. Conversely, desmosomal mutations can affect the electrical activity of the heart by compromising the expression, localization, and/or function of other components of the ‘connexome’, such as NaV1.5, the α subunit of voltage-gated Na+ channels, and Cx43 (see above). Intriguingly, a recent report demonstrated that the Ca2+ cycling machinery is defective in a conditional Pkp2 knockout mouse model [95][33].
Table 1. Lists of genes involved in ACM pathogenesis.
| Gene | Coding Protein | Locus | Reference |
|---|
| desmosomal mutations | JUP | Junction Plakoglobin | 17q21.2 | [35] | [34] |
| DSP | Desmoplakin | 6p24.3 | [36] | [35] | |
| PKP2 | Plakophilin 2 | 12p11.21 | [37] | [36] | |
| DSG2 | Desmoglein 2 | 18q12.1 | [38,39] | [37][14] | |
| DSC2 | Desmocollin 2 | 18q12.1 | [40] | [38] | |
| non-desmosomal mutations | TMEM43 | Transmembrane protein 43 | 3p25.1 | [41] | [39] |
| LMNA | Lamin A/C | 1q22 | [42] | [40] | |
| DES | Desmin | 2q35 | [43] | [41] | |
| CTNNA3 | Alpha-T-catenin | 10q21.3 | [44] | [42] | |
| PLN | Phospholamban | 6q22.31 | [45] | [43] | |
| TGFB3 | Transforming growth factor-3 | 14q24.3 | [27,46] | [44][45] | |
| TTN | Titin | 2q31.2 | [47] | [46] | |
| SCN5A | Sodium voltage gated channel alpha subunit 5 (NaV 1.5) | 3p22.2 | [48] | [47] | |
| CDH2 | Cadherin C | 18q12.1 | [49] | [48] | |
| RYR2 | Type 2 ryanodine receptor | 1q42-q43 | [50,51] | [49][50] |
PKP2
PKP2, encoding for plakophilin-2, is the most frequently mutated gene in ACM patients [37][36]. It has also been linked to other inherited cardiac arrhythmia syndromes, such as Brugada syndrome [96][51], idiopathic ventricular fibrillation, hypertrophic cardiomyopathy, and dilated cardiomyopathy, even if with a low frequency [97][52]. PKP2 is expressed in both myocytes and non-myocytes [54][53]. The main function of PKP2 is to guarantee mechanical stability during the desmosomal intermediate filament assembly required for cell-to-cell contact. PKP2 is part of the so-called ‘connexome’ [98][54]. Different studies highlighted the consequent novel role for PKP2 in the intracellular signaling regulation, electrophysiological and trafficking regulation, and the control of transcription processes [95][33].
Data from a cardiomyocyte-specific tamoxifen-activated Pkp2 homozygous knockout (Pkp2-cKO) mouse model correlated the lack of PKP2 with transcriptional alteration of Ca2+ homeostasis [95][33]. This investigation reported the development of a cardiomyopathy of RV predominance that become evident at 21 days and then progressed into biventricular cardiomyopathy and HF. Transcriptome analysis showed that the transcripts of proteins involved in maintaining the intracellular Ca2+ concentration were downregulated in Pkp2-cKO hearts. The low level of transcripts relevant to Ca2+ cycling, i.e., Ryr2, Ank2, Cacna1c, and Trdn, accompanied a decreased expression of corresponding proteins and the impairment of the EC mechanism [95][33]. Accordingly, the downregulation of AnkB (encoded by Ank2) and triadin (encoded by Trdn), which contribute to maintaining the structural integrity of dyadic junctions [99[55][56],100], significantly reduced the distance between CaV1.2 and RYR2 [95][33]. In addition, Pkp2-cKO-derived cardiomyocytes showed decreased ICa,L density and a slower rate of current inactivation, which is consistent with the reduced expression of Cacna1c [95][33]. It should, however, be pointed out that, although the peak Ca2+ current was decreased, the total Ca2+ charge (i.e., the total amount of Ca2+ entering the cell upon membrane depolarization) was unaltered as compared to wild-type cardiomyocytes because of the slower inactivation rate [95][33]. Notably, the loss of PKP2 was also correlated with a reduction in the SR Ca2+ leak due to the downregulation of both Ryr2 and Casq2 [95][33] (Figure 1). As a consequence, the SR Ca2+ content was remarkably increased in Pkp2-cKO cardiomyocytes, which exhibited an increase in the amplitude and frequency of spontaneous Ca2+ release events (due to RYR2 sensitivity to intraluminal Ca2+ levels) and were therefore more prone to release SR Ca2+ during the EC coupling [95][33]. Accordingly, when Pkp2-cKO cardiomyocytes were paced at increasing rates, they displayed both early and delayed after-transients, which were sufficient to generate ventricular arrhythmogenic events during β-adrenergic stimulation with isoproterenol [95][33]. A more recent investigation focused on the early events driving the remodeling of the Ca2+ handling machinery in RV-derived PKP2-cKO (Pkp2-cKO-RV) cardiomyocytes isolated 14 days after tamoxifen injection [101][57], i.e., when cardiomyopathy was not evident yet. This report revealed an increase in RyR2-dependent Ca2+ release and RyR2-mediated Ca2+ sparks due to the remarkable elevation in the SR Ca2+ load that was caused by a Cx43-dependent increase in membrane permeability [101][57]. Notably, uncoupled Cx43 hemichannels may provide an alternative pathway for extracellular Ca2+ influx [102,103,104][58][59][60] and may, therefore, contribute to refilling the SR Ca2+ store in a SERCA2A-dependent manner. In addition, RyR2’s eagerness to release Ca2+ may be boosted by the observed phosphorylation in Thr2809, an amino acid residue near the consensus sequence for CaMKII and PKA [101][57]. These alterations were not detected in PKP2-cKO LV cardiomyocytes, thereby suggesting that this asymmetric dysregulation of the Ca2+ handling machinery precedes overt ultrastructural alterations and manifestations of ACM.
The relationship between PKP2 and Ca2+ machinery has also been highlighted by a recent bioinformatic approach that took advantage of a database containing transcriptomic information from human hearts, searching for coordinated transcription networks that are subjected to variations based on PKP2 abundance. The results were then validated with the information deriving from Pkp2-cKO murine hearts, thereby confirming the downregulation of RYR2, ANK2, and CACNA1C [105][61]. The results of the combined data supported the idea of a correlation between the PKP2 expression and the abundance of transcripts related to intracellular Ca2+ homeostasis. In this context, mathematical modeling confirmed that the PKP2-dependent downregulation of RYR2 and CASQ2 proteins is sufficient to cause the decrease in SR Ca2+ leak, which results in enhanced SR Ca2+ loading and EC coupling [95][33]. Accordingly, studies carried out on human iPSC-CM from a PKP2-mutated ACM patient showed that they present an abnormal Ca2+ handling capacity [106][62]. Of note, a recent investigation in iPSC-CM suggested that the Ca2+ handling machinery could also be affected by DSG2 mutations. Accordingly, although the systolic and diastolic Ca2+ levels were similar, human ACM iPSC-CM exhibited spontaneous SR Ca2+ release and DADs in both the absence and the presence of β-adrenergic stimulation [107][63]. This investigation did not evaluate the molecular expression of Ca2+-related proteins, but further supports the notion that desmosomal mutations may affect the cardiac Ca2+ toolkit in ACM.
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133.
- Thiene, G.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy: An update. Cardiovasc. Pathol. 2001, 10, 109–117.
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802.
- Bartoli, F.; Sabourin, J. Cardiac Remodeling and Disease: Current Understanding of STIM1/Orai1-Mediated Store-Operated Ca2+ Entry in Cardiac Function and Pathology. Adv. Exp. Med. Biol. 2017, 993, 523–534.
- Xie, W.; Santulli, G.; Guo, X.; Gao, M.; Chen, B.X.; Marks, A.R. Imaging atrial arrhythmic intracellular calcium in intact heart. J. Mol. Cell. Cardiol. 2013, 64, 120–123.
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993.
- Ter Keurs, H.E.; Boyden, P.A. Calcium and arrhythmogenesis. Physiol. Rev. 2007, 87, 457–506.
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561–575.
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- KC, S.; Nair, R.S.; Banerji, A.; Somasundaram, V.; Srinivas, P. Structure activity relationship of plumbagin in BRCA1 related cancer cells. Mol. Carcinog. 2013, 52, 392–403.
- Vangheluwe, P.; Sipido, K.R.; Raeymaekers, L.; Wuytack, F. New perspectives on the role of SERCA2’s Ca2+ affinity in cardiac function. Biochim. Et Biophys. Acta 2006, 1763, 1216–1228.
- van Opbergen, C.J.; Delmar, M.; van Veen, T.A. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: Focus on calcium sensitive pathways. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2017, 25, 157–169.
- Bovo, E.; Huke, S.; Blatter, L.A.; Zima, A.V. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16.
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179.
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021.
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 2005, 96, 346–354.
- Li, J.; Levin, M.D.; Xiong, Y.; Petrenko, N.; Patel, V.V.; Radice, G.L. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J. Mol. Cell. Cardiol. 2008, 44, 597–606.
- Sheikh, F.; Ross, R.S.; Chen, J. Cell-cell connection to cardiac disease. Trends Cardiovasc. Med. 2009, 19, 182–190.
- Garrod, D.R.; Berika, M.Y.; Bardsley, W.F.; Holmes, D.; Tabernero, L. Hyper-adhesion in desmosomes: Its regulation in wound healing and possible relationship to cadherin crystal structure. J. Cell Sci. 2005, 118, 5743–5754.
- Kimura, T.E.; Merritt, A.J.; Garrod, D.R. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J. Investig. Dermatol. 2007, 127, 775–781.
- Wallis, S.; Lloyd, S.; Wise, I.; Ireland, G.; Fleming, T.P.; Garrod, D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol. Biol. Cell 2000, 11, 1077–1092.
- Hennings, H.; Holbrook, K.A. Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp. Cell Res. 1983, 143, 127–142.
- Kartenbeck, J.; Schmid, E.; Franke, W.W.; Geiger, B. Different modes of internalization of proteins associated with adhaerens junctions and desmosomes: Experimental separation of lateral contacts induces endocytosis of desmosomal plaque material. Embo J. 1982, 1, 725–732.
- Mattey, D.L.; Garrod, D.R. Splitting and internalization of the desmosomes of cultured kidney epithelial cells by reduction in calcium concentration. J. Cell Sci. 1986, 85, 113–124.
- Nollet, F.; Kools, P.; van Roy, F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J. Mol. Biol. 2000, 299, 551–572.
- Allen, T.D.; Potten, C.S. Desmosomal form, fate, and function in mammalian epidermis. J. Ultrastruct. Res. 1975, 51, 94–105.
- McHarg, S.; Hopkins, G.; Lim, L.; Garrod, D. Down-regulation of desmosomes in cultured cells: The roles of PKC, microtubules and lysosomal/proteasomal degradation. PLoS ONE 2014, 9, e108570.
- He, W.; Cowin, P.; Stokes, D.L. Untangling desmosomal knots with electron tomography. Science 2003, 302, 109–113.
- Thomason, H.A.; Cooper, N.H.; Ansell, D.M.; Chiu, M.; Merrit, A.J.; Hardman, M.J.; Garrod, D.R. Direct evidence that PKCalpha positively regulates wound re-epithelialization: Correlation with changes in desmosomal adhesiveness. J. Pathol. 2012, 227, 346–356.
- Ohno, S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J. Arrhythmia 2016, 32, 398–403.
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019.
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766.
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106.
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973.
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206.
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164.
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142.
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984.
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821.
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136.
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607.
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210.
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207.
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398.
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thiene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum. Mol. Genet. 1994, 3, 959–962.
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885.
- Erkapic, D.; Neumann, T.; Schmitt, J.; Sperzel, J.; Berkowitsch, A.; Kuniss, M.; Hamm, C.W.; Pitschner, H.F. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace 2008, 10, 884–887.
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605.
- Rampazzo, A.; Nava, A.; Erne, P.; Eberhard, M.; Vian, E.; Slomp, P.; Tiso, N.; Thiene, G.; Danieli, G.A. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum. Mol. Genet. 1995, 4, 2151–2154.
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194.
- Campuzano, O.; Fernandez-Falgueras, A.; Iglesias, A.; Brugada, R. Brugada Syndrome and PKP2: Evidences and uncertainties. Int. J. Cardiol. 2016, 214, 403–405.
- Novelli, V.; Malkani, K.; Cerrone, M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018, 5, 184.
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846.
- Agullo-Pascual, E.; Reid, D.A.; Keegan, S.; Sidhu, M.; Fenyo, D.; Rothenberg, E.; Delmar, M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc. Res. 2013, 100, 231–240.
- Camors, E.; Mohler, P.J.; Bers, D.M.; Despa, S. Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. 2012, 52, 1240–1248.
- Chopra, N.; Knollmann, B.C. Triadin regulates cardiac muscle couplon structure and microdomain Ca2+ signalling: A path towards ventricular arrhythmias. Cardiovasc. Res. 2013, 98, 187–191.
- Kim, J.C.; Perez-Hernandez Duran, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca2+i Homeostasis and Cx43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in PKP2-Deficient Mice. Circulation 2019.
- De Bock, M.; Wang, N.; Bol, M.; Decrock, E.; Ponsaerts, R.; Bultynck, G.; Dupont, G.; Leybaert, L. Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 2012, 287, 12250–12266.
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309.
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480.
- Montnach, J.; Agullo-Pascual, E.; Tadros, R.; Bezzina, C.R.; Delmar, M. Bioinformatic analysis of a plakophilin-2-dependent transcription network: Implications for the mechanisms of arrhythmogenic right ventricular cardiomyopathy in humans and in boxer dogs. EP. Eur. 2018, 20, iii125–iii132.
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110.
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. EP Eur. 2018, 20, f46–f56.
More