One-dimensional (1D) sliding of DNA-binding proteins has been observed by numerous kinetic studies. It appears that many of these sliding events play important roles in a wide range of biological processes. However, one challenge is to determine the physiological relevance of these motions in the context of the protein’s biological function. Here, we discuss methods of measuring protein 1D sliding by highlighting the single-molecule approaches that are capable of visualizing particle movement in real time. We also present recent fifindings that show how protein sliding contributes to function.
- DNA-binding protein
- one-dimensional sliding
- single molecule
1. Introduction
The interactions between proteins and DNA are involved in most cellular functions of DNA, including DNA replication, DNA repair and recombination, and gene transcription and regulation. Many if not all protein–DNA interactions are essential for those processes. When the physical properties of genomic DNA have been well described by a worm-like chain model [1][2][1,2], a number of biophysical studies have provided evidence of protein performing one-dimensional (1D) sliding along the DNA [3][4][5][6][3,4,5,6]. These highly dynamic protein–DNA interactions have served as models for understanding the macromolecular interactions in biology.
The concept of protein 1D sliding has been recognized for several decades. DNA/RNA polymerases and helicases that operate one nucleotide after another are early examples that obviously must move in 1D along a DNA strand [7][8][7,8]. Further investigations have found that many sequence-specific DNA-binding proteins, including a number of restriction endonucleases and transcription factors, also slide along the double-stranded DNA (dsDNA) to search for target sites [9][10][11][9,10,11]. While most studies have inspected protein 1D sliding using relatively simple systems, many complicated biological questions involve protein complex assembly and multiple reaction steps that could not be assessed with these systems. In this review, we have summarized recent studies of protein 1D sliding. Several modes of the sliding and the methods for measuring protein movement have been introduced and discussed here. Of particular interest, we highlight the observations that have ultimately revealed the biological functions of protein 1D sliding.
2. Modes of Protein 1D Sliding
The mechanisms of protein 1D sliding can be categorized into several modes. Probably the most familiar one for biologists is translocation. The majority of DNA/RNA polymerases and helicases hydrolyze nucleoside triphosphate (NTP) and have been suggested to use the released energy for moving along DNA in a unidirectional manner [12][13][14][15][12,13,14,15]. This unidirectional movement is defined as translocation ( Figure 1 a). Moreover, there are other proteins that also translocate along the DNA by NTP hydrolysis but do not couple the directional motions to the nucleotide incorporation or strand separation activity [16]. Those enzymes are referred as translocases. As it can be seen, the translocation is an active motion, and the proteins that perform translocation may be considered as molecular motors on DNA [17][18][17,18]. While the protein translocation activity certainly requires NTP hydrolysis, the reverse is not necessarily true—a protein that performs 1D sliding and contains NTPase activity may not translocate along the DNA (see Section 2.3 ).
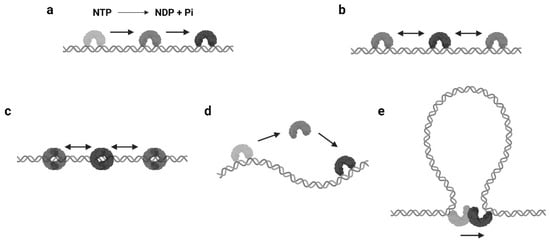
The continuous protein–DNA contacts without dissociation are usually necessary to support facilitated diffusion. However, there are proteins that obviously slide with intermittent DNA contacts. Members of this group include proteins that can form special conformations on DNA, such as a clamp encircling the DNA ( Figure 1 c). A classic example is the DNA replication factor (β-clamp in prokaryotes, also known as proliferating cell nuclear antigen, or PCNA, in eukaryotes), which is a ring-shaped clamp and requires a clamp loader to load it onto the DNA [19][20][22,23]. Coincidentally, the DNA mismatch repair (MMR) proteins, MutS and MutL homologs, also form ATP-dependent ring-like clamps during MMR. These sliding clamps do not always interact with the DNA backbone but can maintain a relatively long lifetime (several minutes) during sliding [21][22][23][24][24,25,26,27]. It is now well established that the MutS and MutL sliding clamps are actually molecular switches that undergo Brownian motion along the DNA, while their ring-like structures prevent the proteins from disassociating from the DNA [21][24][24,27].
Another type of protein sliding with intermittent DNA contacts is hopping/jumping, where the protein diffuses with a series of disassociation and rebinding events [3] ( Figure 1 d). Compared with the facilitated diffusion, one could imagine the protein may slide faster and is able to bypass obstacles during hopping/jumping. As a result, proteins combining facilitated diffusion and hopping/jumping seem to be more efficient when searching for target sites [25][28]. However, distinct from the sliding clamps that are topologically constrained to the DNA, it is now still unclear how the hopping/jumping occurs.
In some scenarios, large DNA molecules can form transient DNA loops. A few proteins are capable of moving between two sites on DNA via these intermediate “loops”, which is referred to as intersegmental transfer ( Figure 1 e). Intersegmental transfer allows the protein to move from one site of DNA to another without dissociation, thus facilitating the target search [3]. However, intersegmental transfer is more likely to occur when the protein contains two or more DNA binding surfaces, since the two binding sites need to be accessed concurrently [3]. Therefore, this mode of sliding only relates to distant translocations on large DNA molecules by a limited subset of DNA-binding proteins [26][27][29,30].
3. Approaches for Observing Protein 1D Sliding
In a typical single-molecule tracking experiment, the DNA is usually immobilized on the quartz surface via the biotin–neutravidin–biotin or digoxigenin– antibody interactions ( Figure 2 a). DNA molecules can be stretched by laminar flow and linked to the surface at both ends, or they can be linked at a single end and stretched by a continuous laminar flow. A similar method such as “DNA curtains”, which uses a combination of nanofabricated surface patterns and fluid lipid bilayers to align hundreds of DNA molecules ( Figure 2 a), has been developed recently [28][35]. However, some single-molecule experiments still suffer from the nonspecific protein binding on the surface, where the undesirable background signals significantly reduce the resolutions of the target proteins. Thus, a novel single-molecule tracking platform named “DNA skybridge” has been designed and applied to monitor proteins only on DNA ( Figure 2 a) [29][36]. Although the quartz barrier used in the DNA skybridge platform has not been commercially available, it is expected that most single-molecule tracking systems can be replaced by this surface-condition-independent and high-throughput method in future. In some single-molecule tracking assays, C-trap optical tweezers combining confocal fluorescence microscope have been used [30][37]. Instead of DNA immobilization on a quartz surface, the C-trap captures a DNA molecule by optical traps ( Figure 2 a), where the changes of DNA lengths and protein positions can be recorded simultaneously. While the C-trap instrument is now commercially available, one disadvantage is that only one molecule can be observed during a single experiment.
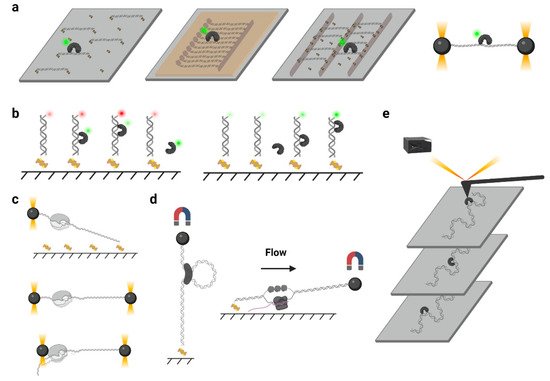
To monitor the DNA–protein interactions in real time, the fluorophore-labeled proteins can be injected into the chamber after DNA immobilization. For single-molecule tracking, the Cyanine (Cy3 and Cy5) and Alexa Fluor dyes, which are small probes that have both high quantum yields and high photostabilities, are recommended. To conjugate the protein with a fluorophore, methods include Cys-maleimide chemistry [31][33], hydrazinyl-iso-pictetspengler ligation [32][38], and sortase-mediated reaction [33][39], which only introduce small tags onto the proteins, and it has been shown that these can largely retain the biological activities of enzymes. In contrast, the commonly used fluorescent proteins are not recommended due to their large sizes and short lifetimes under TIRF illumination [34][40].
Another type of force spectroscopy is referred to as magnetic tweezers, which utilize a similar concept as optical tweezers. Instead of using the optical traps, magnetic tweezers contain magnets that provide an external magnetic field to manipulate dozens of magnetic particles simultaneously. In a general magnetic tweezers set up, a superparamagnetic bead is tethered to one end of a DNA molecule, while the other end of the DNA is attached to the slide surface [35][53] ( Figure 2 d, left). The external magnets are able to impart both twist and tension to the DNA, making magnetic tweezers an excellent method to study the motions of DNA translocases or topoisomerases [36][37][38][54,55,56]. Alternatively, a flow-stretching set-up has been designed, where the instrument combines the magnetic force and the drag force created by a laminar flow to hold the paramagnetic beads and stretch the DNA ( Figure 2 d, right). Using the flow-stretching assay, a number of DNA molecules have been tracked in real time, and the DNA replication, unwinding, and excision events were studied [39][40][41][57,58,59]. Due to the large volume of the magnetic field, the magnetic force spectroscopy is able to monitor hundreds of DNA molecules at one time but with lower spatial and temporal resolutions.
Atomic force microscopy (AFM) generates images by scanning a small cantilever with a sharp tip over the surface of a sample. By placing the tip in contact with the molecule of interest and moving the surface with respect to the tip, force can be modulated precisely, thereby changing the cantilever deflection. Cantilever deflection is further monitored by a laser beam reflected from the sharp tip onto a position-sensing detector [42][60] ( Figure 2 e). High-speed atomic force microscopy (HS-AFM) has been used to image the DNA–protein complex in real time, where the protein diffusion on DNA can be visualized [43][61]. Nevertheless, the main limitations of AFM come from the nonspecific interactions between proteins and the surface that may suppress the sliding [43][61].
4. Biological Significance of Protein 1D Sliding
Protein 1D sliding has been shown to be involved in many biological processes. Here, we summarize and discuss the significance of protein 1D sliding in biology.
One of the well-recognized functions of protein 1D sliding is to accelerate the target search process. For many DNA-binding proteins that specifically recognize particular sequences, such as transcription factors, endonucleases, and DNA methyltransferases, the target sites only constitute a minute fraction of the genome DNA ( Figure 3 a). Thus, the target search should be highly efficient. Actually, it is the case. Some of the proteins locate their target sites much more rapidly than the theoretical calculated one that uses a general diffusion–collision mechanism [44][19]. The search process was later modeled as a two-step event, where nonspecific protein–DNA binding first occurs and is followed by a 1D sliding to the target site. For example, the lacI repressor [44][45][19,20], tumor suppressor p53 [11][46][11,62], and endonucleases EcoRV, BcnI, and Endonuclease V [9][47][48][49][50][9,63,64,65,66] have been reported to slide along the DNA to facilitate the target search. Interestingly, while the continuous protein–DNA contacts without dissociation were initially believed to be essential for the searching processes [11][47][48][11,63,64], further evidence have shown that the sliding was accompanied by occasional hopping/jumping events, which helped the enzymes to locate their target sites [9][46][49][50][51][52][53][54][9,62,65,66,67,68,69,70]. The combination of sliding with continuous DNA contacts and hopping/jumping suggests a trade-off between speed and accuracy during the target search [52][68].
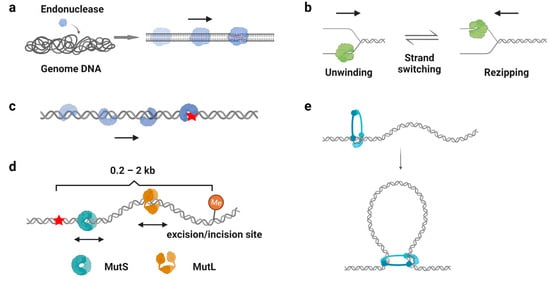
As mentioned above ( Section 2.3 ), β-clamp/PCNA forms a special ring-shaped conformation on DNA that provides a mobile platform to tether the DNA polymerase. While the replication sliding clamp itself usually adopts an intrinsically closed ring-like configuration, a sliding clamp loader belonging to the AAA+ ATPase family first binds ATP and recognizes the β-clamp/PCNA molecule to produce a cleft on the clamp [55][84]. Then, this complex binds a primer–template junction on DNA to stimulate the ATPase activity of the clamp loader [56][57][85,86]. ATP hydrolysis releases the loader from the β-clamp/PCNA and DNA, leading to the closure of the β-clamp/PCNA on substrate DNA [58][59][87,88]. Studies have demonstrated that the PCNA moves along DNA using two different sliding modes: it slides by tracking the DNA backbone in the rotational-coupled mode, while it moves at higher rates in the non-helical diffusion mode [60][61][89,90]. Most importantly, the replication sliding clamp exhibits a half-life of tens of minutes on DNA that enhances polymerase processivity by more than 1000-fold [62][63][91,92]. Other clamp-like proteins that have a long lifetime on DNA, such as the Mre11-Rad50-Nbs1 (MRN) complex, MutS, and MutL homologs, also have been shown to act as processivity factors during DNA repair [40][41][64][65][58,59,93,94].
DNA damage occurs at a rate of 10,000 to 1,000,000 lesions per cell per day [66][95]. To maintain the stability of the genome, repair proteins have to recognize DNA lesions efficiently. Many DNA repair proteins have been reported to scan along the DNA for damage sites ( Figure 3 c), including the base excision repair protein glycosylases [67][68][69][70][96,97,98,99], mismatch repair protein MutS homologs [71][72][41,100], and nucleotide excision repair protein Rad4, XPA, and UvrB [73][74][101,102]. Among these studies, it is generally believed that the 1D sliding is used to follow the DNA backbone for lesion recognition, where a damage site only slightly destabilizes the helical structure of a duplex DNA [71][70][72][41,99,100]. In other words, the scanning processes likely involve rotation-coupled diffusions along the DNA helix, while the proteins are in continuous contact with the duplex DNA [71][75][41,103].