Tuberculosis (TB) is an airborne infectious disease caused by the bacillus Mycobacterium tuberculosis (MTB). To date, MTB still represents one of the most dangerous pathogens, claiming millions of lives each year worldwide. Polypharmacology could have features that make it an approach more effective than the classical polypharmacy, in which different drugs with high affinity for one target are taken together. Firstly, for a compound that has multiple targets, the probability of development of resistance should be considerably reduced. Moreover, such compounds should have higher efficacy, and could show synergic effects. Lastly, the use of a single molecule should be conceivably associated with a lower risk of side effects, and problems of drug–drug interaction.
1. Introduction
Tuberculosis (TB) is an airborne infectious disease caused by the bacillus
Mycobacterium tuberculosis (MTB). To date, MTB still represents one of the most dangerous pathogens, claiming millions of lives each year worldwide. As reported by the WHO Global report of 2019, in 2018 at least 10 million people fell ill with TB, of which 1.2 million died among HIV-negative subjects, but the number grows with an additional 251,000 deaths among HIV-positive patients
[1]. Even if the numbers are still high, there is an overall reduction in recent years, this can be correlated to the improvements in the EndTB Strategy, that started in 2015, whose aim is to reduce by 80% the incidence of TB worldwide and by 90% the number of deaths caused by this single infectious agent before 2030
[1].
Understanding the dynamics of MTB transmission is fundamental to control and prevent TB spreading, especially in high burden countries where this pathogen is still endemic. Genotyping and spoligotyping are established strategies for the molecular characterization and identification of MTB strains
[2]. The complex interactions between MTB and the host during first contact, infection, and persistence are yet to be fully understood. The human body response to infectious agents calls for the mobilization of the innate immune system cells, but MTB can evade the immune response and persist in the human body thanks to its high genome plasticity
[3]. In addition, upon MTB exposure, only a small percentage of patients will develop an active form of TB, while the majority of them will have a latent TB infection (LTBI)
[4]. LTBI is a persistent immune response to MTB antigens in the absence of clinical symptoms of TB and can be diagnosed with the interferon-γ release assay (IGRA) and the tuberculin skin tests (TST), but the diagnosis of LTBI is not predictive of developing active TB
[5]. Generally, LTBI can switch into the active form of TB in case of a strong immune system weakening, such as in case of HIV co-infection, or autoimmune diseases, but it can occur in not-high-risk patients, as well
[5].
Treatment for active drug-susceptible TB is expensive and long, taking up to six months of daily doses of four first line drugs: Isoniazid, rifampicin, ethambutol, and pyrazinamide. Treatment success rate is generally higher when the patient follows the treatment to its completion. Low treatment success can depend on multiple factors, poor counseling during the treatment phase with consequent drop-out, high-risk populations (refugees, poor), co-morbidity with other pathologies
[6]. For these reasons, supporting and informing the patients is pivotal. Furthermore, treatment outcomes depend on the spreading of MTB drug-resistant strains, for which the canonical therapy does not work
[7]. Drug susceptibility tests should be performed, not only before, but also during therapy, to allow for an early identification of developing drug-resistant TB (DR-TB)
[7].
Therefore, DR-TB should be carefully treated based on the resistance phenotype of the MTB drug-resistant strain. As recommended by the WHO consolidated guidelines on drug-resistant tuberculosis treatment of 2019, rifampicin-susceptible and isoniazid-resistant TB should be treated with daily doses of rifampicin, ethambutol, pyrazinamide, and levofloxacin or other fluoroquinolones
[8].
Poor management of DR-TB can develop in multi-drug resistant TB (MDR-TB) with acquired rifampicin-resistance (RR-TB) with or without resistance to other first line drugs. MDR-TB/RR-TB treatment can be highly challenging, usually needing personalized strategies for each patient
[9].
Treatment regimen for MDR-TB requires eight months of daily administration of pyrazinamide in association with at least four more second-line drugs. The duration of the treatment can vary from 12 to 20 months, but mostly depends on the patient response and TB evolution
[9]. In the treatment of MDR-TB, fluoroquinolones, injectable anti-TB drugs, ethionamide, and cycloserine should be used in association to pyrazinamide for the first intensive phase, while pyrazinamide should be continued for the entirety of the treatment
[9]. If cycloserine cannot be used, the
para-aminosalicylic acid (PAS) should be used instead
[9]. Streptomycin should be used as a second-line drug only for amikacin-resistant MDR-TB, as it correlates with a reduced treatment success rate
[8].
The spreading of MDR MTB strains has become a major health problem worldwide, especially in high burden countries, where in depth analysis, exhaustive follow-up information retrieval, and organic procedures are challenging
[10]. Furthermore, due to poor management of treatment administration and overall inadequate antibiotic distribution, MDR-TB can dramatically evolve into extensively drug resistant TB (XDR-TB) resistant to isoniazid and rifampicin, associated with resistance to at least one fluoroquinolone and one injectable second line drug
[11].
2. How Polypharmacology Can Help in Fighting MDR-TB?
Drug resistance mechanisms are a main problem to face in fighting MDR- and XDR-TB strains. A pathogen, indeed, requires often just a single base mutation to became resilient to antibiotics or chemotherapeutics. The development of a single pharmaceutical molecule that acts on different targets is a possible strategy to bypass this problem. This multitargeting system is known as “polypharmacology”. SQ109, for example, is a promising multitarget drug that putatively hits four different targets in the respiratory chain of
M. tuberculosis [12].
Polypharmacology is in contraposition with the more classical “polypharmacy”, the use of different drugs, each with high affinity for one target, that are taken together as cocktails or multicomponent drugs. Even if both strategies are effective, polypharmacology possesses important features to underline. Firstly, the use of only one molecule instead of many is conceivably associated with a possible lower cytotoxicity and side effects. Moreover, polypharmacology is expected to have a higher therapeutic efficacy, compared to the classical approach of hitting only one best target at a time
[13]. Moreover, multitarget drugs show synergic or additive effects, this means that they can modulate complex diseases in lower time and with smaller doses thanks to simultaneous targeting
[14]. Lastly, they may avoid the problem of drug–drug interaction. All these features allow the consequential improvement of the patient quality of life
[15].
For these reasons, polypharmacology is an emerging strategy in therapeutic development of drugs against synergistic bacterial diseases
[16], neurological diseases
[17], and cancer
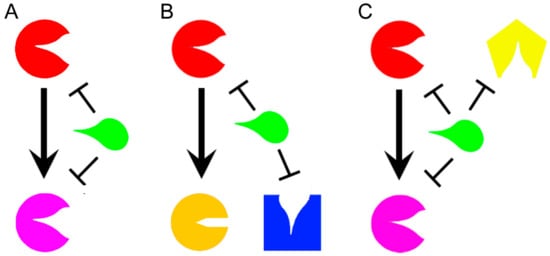
[18]. Even if the development of multitarget drugs is a relatively novel field, some classifications have been already proposed according to their mechanisms of action, or their structures. Based on mechanisms of action, a molecule that affects different targets within the same metabolic pathway acts in “vertical targeting”. We can distinguish a “series inhibition” if the two targets are related (
Figure 1A), as for example consequential or in the same pathway, while a “parallel inhibition” (
Figure 1B) when they are unrelated, but should have for example a common substrate that can be mimicked
[12]. The vertical targeting is a strategy that can fight the insurgence of certain kinds of resistance mechanisms such as mutations
[19]. A drug that acts in “network targeting” instead, hits different targets in different pathways (
Figure 1C)
[12], and it is able to prevent compensatory homeostatic responses and the adaptive resistance
[14].
Figure 1. Representation of a general drug (green) that acts in vertical targeting in series (A) when it inhibits two related enzymes or in parallel (B) when it targets two unrelated enzymes with a common substrate. A drug acts in network inhibition (C) when it targets different enzymes in different metabolic pathways. The different targeted enzymes are arbitrary represented in different colors (Red, Pink, Yellow, Orange, and Blue).
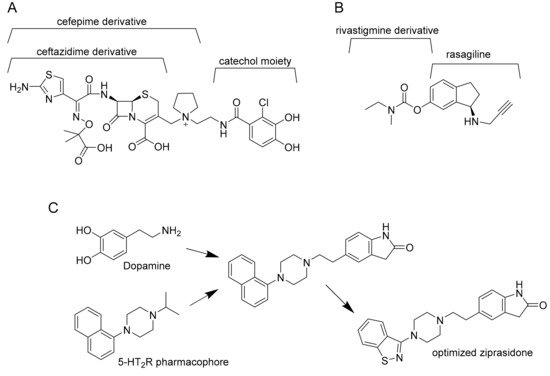
By structure, multitarget drugs may be classified according to the optimization and modifications that are introduced by chemical tailoring to the original molecule. Another possibility is to rationally design a multitarget drug by virtual studies. This strategy gives rise to three possibilities: Linked, fused, or merged pharmacophores (
Figure 2).
Figure 2. Example of linked (A), fused (B), and merged pharmacophores (C). Cefepime derivative is the linker between ceftazidime derivative and catechol moiety in cefiderocol (A). Ladostigil, an analogue of compound 9, is derived from the fusion of rasagiline with rivastigmine (B). Finally, dopamine is merged with 5-HT2R pharmacophore and the obtained molecule is optimized to ziprasidone (C).
Linked pharmacophores are molecules bound together by a stable or biodegradable linker
[20]. The result is a larger molecule that does not need further improvement, but the position of the linker is crucial for its final effect on target. Moreover, this molecule may fail to reach the intracellular compartment of interest because of its size. From another point of view, some linkers can improve solubility and polarity of the molecule and reduce unspecific diffusion into an unwanted cellular compartment
[21]. Nonetheless, linked pharmacophores are currently used to produce antibody drug conjugates, that is, a drug (usually a small molecule) conjugated with an antibody that works as a vessel to reach the target. Another example is to conjugate a drug, as for example an antibiotic, to bacterial siderophores. The resulting sideromycin exploits the bacterial uptake mechanisms to be internalized. A promising example is Cefiderocol. This compound, that binds the penicillin-binding protein 3 (PBP3) inhibiting cell wall biosynthesis, is active against different multidrug-resistant Gram-negative pathogens such as carbapenem-resistant
Pseudomonas aeruginosa, Acinetobacter baumannii, and
Enterobacteriaceae, and is currently in phase II clinical trials
[22].
Fused pharmacophores are the result of two joint small molecules without a linker. According to the type of final bond (imine, ester, or hydrazine bonds) they may be cleavable or not. Moreover, the position of the bond is crucial for the final effect of both molecules and the resulting pharmacophore may be a large molecule with all the consequent problems for its delivery. According to Sterling et al.
[23], for example, the “compound 9” described in their article is active against both acetylcholinesterase (AChE) and monoamine oxidases (MAOs), and it was created by fusion of rivastigmine and rasagiline
[23].
Finally, merged pharmacophores are possible only when a part of two different molecules can overlap, resulting in their integration. Anyway, to be active, the resulting molecule must possess the correct molecular geometry and charge distribution to interact with both targets
[24]. For example, Ziprasidone is an antipsychotic drug optimized by the resulting fusion of dopamine and 5-HT
2R pharmacophore. It targets both type 2 dopamine receptors D2 and type 2 serotonin synergic receptors 5-HT
2R
[25].
3. Multitargeting Compounds Against M. tuberculosis
The single-target strategy for the development of novel antimicrobial drugs involves single proteins essential for survival of the pathogen. However, this approach shows a weakness, since a single mutation in the target protein could be sufficient to confer resistance. For this reason, drug combinations in TB treatment are preferred over single-drug therapies; in this context multitarget drug discovery may offer a novel opportunity
[12]. In recent years, several multitargeting antitubercular compounds have emerged
[26]. Many compounds have been firstly discovered through phenotypic screening, then they were retrospectively found to target multiple proteins simultaneously, such as the case of the MmpL3 inhibitors
[12]. Interestingly, in some cases the compound has been designed or selected as an inhibitor of a specific enzyme or pathway, then it was found to have additional targets. For instance, the recently described 5-(5-nitrothiophen-2-yl)-4,5-dihydro-1
H-pyrazoles, have been selected as potential inhibitors of the arylamine
N-acetyltransferase enzyme, and were found to be also potent efflux pump inhibitors
[27][28][27,28]. Another example is the case of the tetrahydroisoquinoline compounds, that have been selected as inhibitors of the ATP-dependent MurE ligase, but were found to have pleiotropic mechanisms of action, not fully clarified yet
[29]. On the other hand, compounds are also emerging from biochemical screens against specific targets, as for instance the GroEL/ES chaperonin and protein tyrosine phosphatase B inhibitors
[30]. Finally, multitargeting compounds have been specifically designed, as for instance the fatty acids bypass biosynthetic pathways inhibitors
[31]. The most recent and significant examples of the different multitargeting antitubercular compounds are here described.