The human microbiome is defined as the full array of the diverse microorganisms (microbiota) that live on and in humans, as well as their genetic materials. It is considered one of the leading environmental factors in disease development, with Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria dominant species. Human microbiota manifestation is influenced by multiple environmental and physiological changes, including age, sex, race, geography, diet, host genetics and lifestyle, drugs like antibiotics, and interaction with the immune system and metabolic pathway.
- metabolomics
- microbiome
- microenvironment
- breast cancer
1. Introduction
Breast cancer is a heterogeneous disease and one of the world’s most prevalent malignancies. According to the World Health Organization (WHO), 2.3 million women were diagnosed with breast cancer in 2020, with 685,000 deaths globally ( https://www.who.int/news-room/fact-sheets/detail/breast-cancer , accessed on 15 September 2021). Lately, Breast cancer incidence has increased to 29.7% among Saudi women [1]. Several risk factors are associated with breast cancer; they are mainly classified as modifiable and non-modifiable factors. Non-modifiable risk factors include age, menopause, family history, hormonal variations, and genetic susceptibility. Modifiable risk factors such as diet, lack of physical activity, obesity, alcohol consumption, and oral contraceptive can be changed if appropriate measures are taken [1][2][3][1,2,3]. Normally, patients develop breast-related signs such as lumps, size alteration, pains, and nipple fluid discharge [4]. Although women are at a higher risk of developing breast cancer than men, breast cancer may occur in males, who represent less than 1% of overall breast cancer cases [5]. Despite the low incidence of breast cancer in males, the mortality rate is considered high as the disease is often only discovered at the final stage. Compared to females, breast tumors in males are more often of the ductal carcinoma type and estrogen- and progesterone-receptor positive [6].
There are five main molecular subtypes of breast cancer that are associated with the expression of three receptors in tumor cells, namely estrogen (ER), progesterone (PR), and human epidermal growth factor receptor-2 (HER2) ; Luminal A cancers largely correspond to ER or PR positive, HER2 negative, and low histological grade/proliferation rate, while Luminal B tumors display relatively lower levels of ER or PR expression, and either exhibit HER2 amplification, high histological grade/high proliferation, or both. The HER2-enriched group (ERBB2) consists of ER-negative tumors and expresses genes mapping to the HER2 amplicon. Additionally, triple-negative breast cancer phenotype (TNBC) is formed by basal-like cancers characterized by low or /lacking levels of expression of ER and ER-related genes (including PR) and the frequent absence of HER2 overexpression. Normal breast-like subtype tumors show remarkable similarities with normal breast and fibroadenomas samples at the messenger Ribonucleic Acid (mRNA) expression level. There are three histological grades: grade 1—well-differentiated; grade 2—moderately differentiated; and grade 3—poorly differentiated [7][8][9][7,8,9].
Tumor microenvironment cells (TME) play a crucial role in cancer development and progression [10]. The heterogeneity of the TME mainly consists of the extracellular matrix (ECM) and various types of tumor stromal cells, including immune and inflammatory cells, endothelial cells, adipocytes, bone marrow-derived cells, and fibroblasts [11]. Endothelial cells are critical to the development of tumor angiogenesis, which provides metastatic tumor cells entry to the circulatory system [12]. Fibroblast cells are considered one of the most abundant and significant types of cells in the TME. Normally, fibroblasts play a key role in wound healing, epithelial differentiation regulation, and inflammation [13][14][13,14]. However, they are present in either activate or inactivated forms inside tumors, commonly known as cancer-associated fibroblast (CAF) or/myofibroblasts [13]. In cancer, CAFs trigger invasion, progression, and metastasis [15].
Recently, researchers has shown great interest in understanding and connecting the inflammation mechanism involved in breast cancer with the breast tissue microbiome [2][16][17][2,20,21]. Disturbance of the microbiome has been linked to chronic diseases and malignancies, including breast cancer. Microbial alterations observed in breast cancer highlight the possible role of microbiota in breast cancer development, prevention, and management [3]. Microbiome expression is associated with the excreted metabolome, which helps study the disease phenotypes and develop biomarkers for disease management. This rentryview introduces updated literature on the connection between the TME and breast cancer development, and discusses the association between the tissue microbiome and metabolic changes in disease development.
2. Methods for Studying the Microbiota
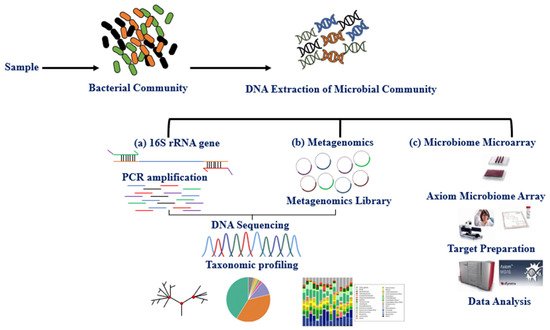
The microbiota can be studied directly using traditional culture-dependent or molecular approaches and indirectly through its association with other biomolecules or omics approaches such as epigenetics and metabolomics. The primary molecular technique for studying microbiota expression is DNA amplification of hypervariable regions using polymerase chain reaction (PCR). Microbiota identification (sequencing) and expression level are obtained using next-generation sequencing technologies (NGS) and microarray [18][35]. Multiple studies have explored the variable regions (V1–V9) 16S rRNA, shared by bacteria and archaea, using the NGS, whole-genome shotgun sequencing (WGS), and DNA microarray (e.g., PathoChip) techniques [19][20][21][36,37,38]. These techniques together have contributed to the study of the human microbiome and established an association between imbalance in the microbiome (dysbiosis) and disease phenotypes [22][23][24][25][39,40,41,42]. The International Human Microbiome Standard ( www.microbiome-standards.org , accessed on 25 August 2021) and the Microbiome Quality Control project ( www.mbqc.org , accessed on 25 August 2021), have developed standard operating procedures (SOP) designed to improve data quality and comparability in the human microbiome field ( Figure 1 ) [21][38].
3. Breast Tissue Microbiome
The microbiota ’s dysbiosis has contributed significantly to breast cancer progression, and other health conditions, as reviewed elsewhere [17][26][21,24]. Microbes may, directly or indirectly, influence the development of breast cancer. The direct effect involves microbes on skin/breast tissue that contribute to breast cancer progression via contact with breast tissue. On the other hand, the indirect effect involves structural and functional components of bacteria, secretion products (e.g., quorum sensing peptides), or bacterial metabolites [17][26][27][28][21,24,34,43]. Several studies that describe the correlation between tissue microbiome dysbiosis and breast cancer development have revealed distinct species expression in patients compared to healthy individuals ( Table 1 ) [29][30][44,45]. Urbaniak et al. [16][20] reported a higher abundance of Prevotella , Lactococcus , Streptococcus , Corynebacterium , and Micrococcus in healthy women, while Bacillus , Staphylococcus , Enterobacteriaceae , Comamondaceae , and Bacteroidetes were more abundant in women with breast cancer. In a more comprehensive study based on tissue samples collected from The Cancer Genome Atlas (TCGA), Mycobacterium fortuitum , and Mycobacterium phlei were found abundant in breast cancer tissues ( n = 668) compared to the normal adjacent tissues ( n = 72) [31][46]. Another study offered substantial evidence connecting breast cancer development to microbiome diversity and expression, where Methylobacterium growth was significantly decreased in cancer patient breast tissues [27][34]. A previous study reported a relative increase of Methylobacterium radiotolerans in tumor tissue versus Sphingomonas yanoikuyae in healthy adjacent tissue. The bacterial DNA load showed an inverse correlation with the stage of breast cancer disease [2][32][2,47]. Therefore, bacterial load may be linked to reduced gene expression of the antibacterial response gene in advanced-stage breast cancer [2]. Costantini et al. [30][45] studied the multi-hypervariable region of the 16S-rRNA gene and found that the V3 region is the most informative for breast tissue microbiota. The microbiota imbalance may lead to downstream malfunction of the immune system, permitting tumor development [33][19]. Of note, most of these studies have sequenced the 16S rRNA gene using qPCR, NGS, or DNA microarray (PathoChip) methods for bacterial identification, as summarized in Table 1 .
| Sample Type and Size | Method | Variable Region | Changes to the Microbiome | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Healthy | Benign | Cancer | Adjacent | Healthy Patients | Cancer Patients | Adjacent | |||
| 20 | 20 | NGS | V4 | ↑ Methylobacterium radiotolerans | ↑ Sphingomonas yanoikuyae | [2] | |||
| 24 | 17 | 22 | NGS | V3–V4 | ↓ Methylobacterium | [27][34] | |||
| 23 | 13 | 45 | NGS | V6 | ↑ Prevotella, Lactococcus, Streptococcus, Corynebacterium, and Micrococcus | ↑ Bacillus, Staphylococcus, Enterobacteriaceae, Comamondaceae, and Bacteroidetes. | [16][20] | ||
| 668 | 72 | NGS | V3–V5 | ↑ Mycobacterium fortuitum and Mycobacterium phlei | [31][46] | ||||
| 5, Canadians | 11 | 27 | NGS | V6 | The most abundant taxa in the Canadian samples were: Bacillus (11.4%), Acinetobacter (10.0%), Enterobacteriaceae (8.3%), Pseudomonas (6.5%), Staphylococcus (6.5%), Propionibacterium (5.8%), Comamonadaceae (5.7%), Gammaproteobacteria (5.0%), and Prevotella (5.0%). | [34][48] | |||
| 5, Irish | 33 | The most abundant taxa in the Irish samples were: Enterobacteriaceae (30.8%), Staphylococcus (12.7%), Listeria welshimeri (12.1%), Propionibacterium (10.1%), and Pseudomonas (5.3%). ↑ Escherichia coli |
[34][48] | ||||||
| 20 | 50, BRER 34, BRHR 24, BRTP 40, BRTN |
PathChip array | Unique and common microbial signatures in the major breast cancer types are summarized in Table 1 in (51) All four breast cancer types had dominant signatures for Proteobacteria followed by Firmicutes. Actinomyces signatures were also detected in each breast cancer types. |
[35][49] | |||||
| 9, CNB 7, SEB 3, Both |
9, CNB 7, SEB 3, Both |
NGS | V2–V4 V6–V9 |
Proteobacteria are the most abundant phylum followed by Firmicutes, Actinobacteria, and Bacteroidetes. The presence of the genus Ralstonia is associated with breast tissue. The relative abundance of Methylobacterium was different in certain patients. |
[30][45] | ||||
NGS: Next-generation sequencing, qPCR: quantitative Polymerase chain reaction, BRER: endocrine receptor (estrogen or progesterone receptor) positive, BRHR: human epidermal growth factor receptor 2 (HER2) positive, BRTP: triple positive (estrogen, progesterone, and HER2 receptor-positive), BRTN: triple-negative (absence of estrogen, progesterone, and HER2 receptors), CNBs: core needle biopsies, SEBs: surgical excision biopsies. Up and down arrows refer to up- and down-regulated bacteria, respectively.
4. Interaction between Microbiome and Metabolomics in Breast Cancer
Recently, interest in studying the association between the microbiome and metabolic alteration in cancer has increased. Microbiome metabolites can be critical modulators of the TME by regulating, either positively or negatively, vital processes such as inflammation, proliferation, and cell death [36][79]. However, research investigating the interaction between the microbiome and the metabolome in breast cancer is limited. Only a few reports have highlighted the association between the breast microbiome and metabolome in the breast cancer microenvironment. A previous study reported a higher abundance of Bacillus cereus in breast cancer patients compared with healthy controls. Bacillus cereus metabolizes progesterone into 5-alpha-pregnane-3,20-dione, stimulating cell proliferation and tumor progression [16][37][20,80]. Moreover, dysbiosis of the gut microbiome leads to elevated activities of β-glucuronidase, which is responsible for estrogen reactivation through the deconjugation of conjugated estrogens, and hence, an increased risk of estrogen-related conditions such as breast cancer [38][39][81,82]. A recent LC-MS metabolomics study reported a correlation between the gut microbiome and choline metabolism in breast cancer patients. The lower abundance of Faecalibacterium was linked to the upregulation of phosphocholine levels. [40][83]. The study suggested that combining flora-metabolites with the flora-bacteria (e.g., Faecalibacterium combined with phosphocholine) might serve as promising diagnostic biomarkers for breast cancer, and that Faecalibacterium may suppress breast cancer proliferation and invasion by inhibiting IL-6 signal transducers and activators of the transcription 3 (STAT3) pathway [40][83]. Lithocholic acid is a bacterial metabolite that could influence cancer cell proliferation through activation of the Takeda G-protein-coupled receptor 5 (TGR5) [41][84]. Bacterial metabolites, lithocholic acid, short-chain fatty acids, indole-propionic acid (IPA), or cadaverine can limit the proliferation of breast cancer cells [41][42][43][84,85,86]. These findings suggest that a deeper understanding of the link between microbiome and metabolome in breast cancer may provide new biomarkers as well as, therapeutic and prevention strategies.