Fluorine is widely dispersed in nature and has multiple physiological functions. Although it is usually regarded as an essential trace element for humans, this view is not held universally. Moreover, chronic fluorosis, mainly characterized by skeletal fluorosis, can be induced by long-term excessive fluoride consumption. High concentrations of fluoride in the environment and drinking water are major causes, and patients with skeletal fluorosis mainly present with symptoms of osteosclerosis, osteochondrosis, osteoporosis, and degenerative changes in joint cartilage. Etiologies for skeletal fluorosis have been established, but the specific pathogenesis is inconclusive. Currently, active osteogenesis and accelerated bone turnover are considered critical processes in the progression of skeletal fluorosis. In recent years, researchers have conducted extensive studies in fields of signaling pathways (Wnt/β-catenin, Notch, PI3K/Akt/mTOR, Hedgehog, parathyroid hone, and insulin signaling pathways), stress pathways (oxidative stress and endoplasmic reticulum stress pathways), epigenetics (DNA methylation and non-coding RNAs), and their inter-regulation involved in the pathogenesis of skeletal fluorosis.
- skeletal fluorosis
- fluoride
- endemic disease
- signaling pathways
- epigenetics
- endoplasmic reticulum stress
- oxidative stress
1. Introduction
2. Mechanism of Signaling Pathways in Skeletal Fluorosis
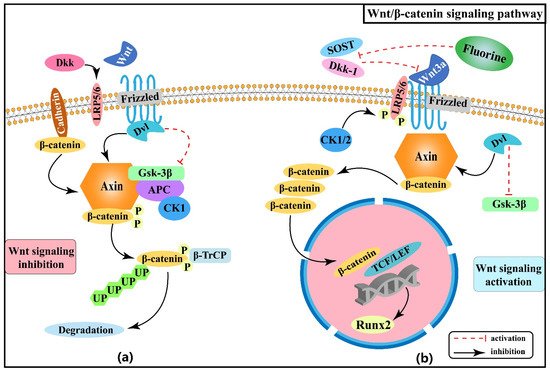
2.1. Effect of Wnt/β-Catenin Signaling Pathway on Skeletal Fluorosis
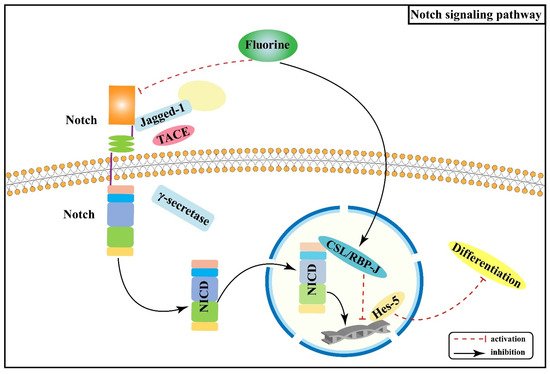
2.2. Effect of Notch Signaling Pathway on Skeletal Fluorosis
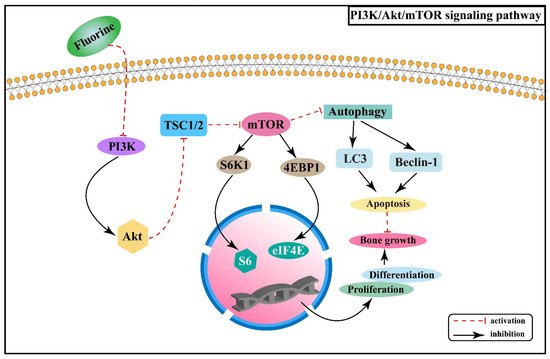
2.3. Effect of PI3K/Akt/mTOR Signaling Pathway on Skeletal Fluorosis
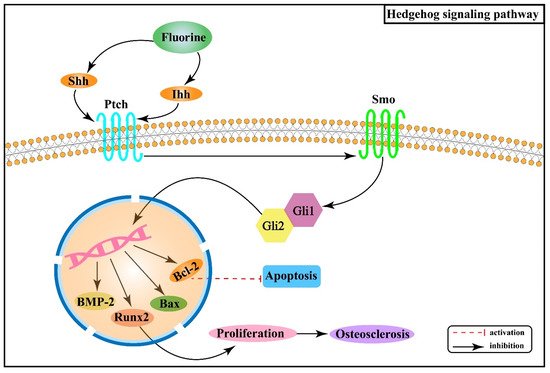
2.4. Effect of Hedgehog Signaling Pathway on Skeletal Fluorosis
2.5. Effect of Hormones and Their Receptor Signaling Pathways on Skeletal Fluorosis
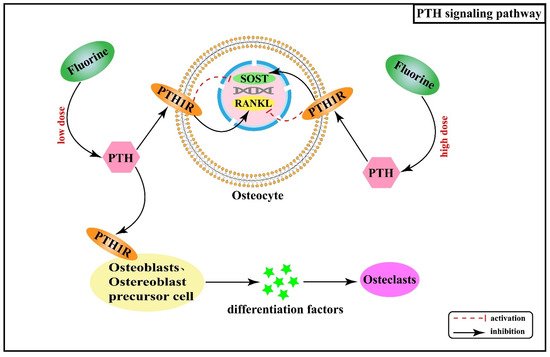
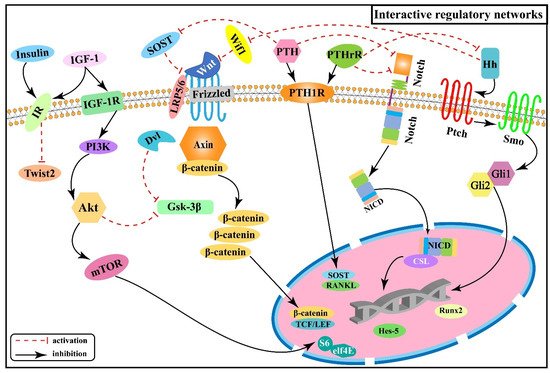
2.6. Interactive Regulatory Networks among Signaling Pathways Involved in Skeletal Fluorosis
3. Mechanism of Stress Pathways in Skeletal Fluorosis
3.1. Effect of Endoplasmic Reticulum Stress on Skeletal Fluorosis
3.2. Effect of Oxidative Stress on Skeletal Fluorosis
4. Mechanism of Epigenetics in Skeletal Fluorosis
4.1. Effect of DNA Methylation on Skeletal Fluorosis
4.2. Effect of Non-Coding RNAs on Skeletal Fluorosis
5. Conclusions
References
- Zhao, Y.Y. The Progress about the influence of Fluorine on Bone. Med. Recapitul. 2006, 12, 1092–1094.
- Srivastava, S.; Flora, S.J.S. Fluoride in Drinking Water and Skeletal Fluorosis: A Review of the Global Impact. Curr. Environ. Health Rep. 2020, 7, 140–146.
- Fan, Z.P.; Gao, Y.H.; Wang, W.; Gong, H.Q.; Guo, M.; Zhao, S.C.; Liu, X.H.; Yu, B.; Sun, D.J. Prevalence of Brick Tea-Type Fluorosis in the Tibet Autonomous Region. J. Epidemiol. 2016, 26, 57–63.
- Izuora, K.; Twombly, J.G.; Whitford, G.M.; Demertzis, J.; Pacifici, R.; Whyte, M.P. Skeletal fluorosis from brewed tea. J. Clin. Endocrinol. Metab. 2011, 96, 2318–2324.
- Ando, M.; Tadano, M.; Asanuma, S.; Tamura, K.; Matsushima, S.; Watanabe, T.; Kondo, T.; Sakurai, S.; Ji, R.D.; Liang, C.K.; et al. Health effects of indoor fluoride pollution from coal burning in China. Environ. Health Perspect. 1998, 106, 239–244.
- Xu, Y.Y.; Huang, H.; Zeng, Q.B.; Yu, C.; Yao, M.L.; Hong, F.; Luo, P.; Pan, X.L.; Zhang, A.H. The effect of elemental content on the risk of dental fluorosis and the exposure of the environment and population to fluoride produced by coal-burning. Environ. Toxicol. Pharmacol. 2017, 56, 329–339.
- Mou, W.P.; Yan, H.; Zhang, L.H. Progress in molecular mechanism of skeletal fluorosis. Chin. Foreign Med. Res. 2011, 9, 158–160.
- Noël, C.; Gosselin, B.; Dracon, M.; Pagniez, D.; Lemaguer, D.; Lemaître, L.; Dhondt, J.L.; Lelièvre, G.; Tacquet, A. Risk of bone disease as a result of fluoride intake in chronic renal insufficiency. Nephrologie 1985, 6, 181–185.
- Wei, W.; Pang, S.J.; Sun, D.J. The pathogenesis of endemic fluorosis: Research progress in the last 5 years. J. Cell Mol. Med. 2019, 23, 2333–2342.
- Boivin, G.; Chavassieux, P.; Chapuy, M.C.; Baud, C.A.; Meunier, P.J. Skeletal fluorosis: Histomorphometric findings. J. Bone Miner. Res. 1990, 5, S185–S189.
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525.
- Day, T.F.; Guo, X.Z.; Garrett-Beal, L.; Yang, Y.Z. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750.
- Patel, M.S.; Karsenty, G. Regulation of bone formation and vision by LRP5. N. Engl. J. Med. 2002, 346, 1572–1574.
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192.
- Chu, Y.R.; Gao, Y.H.; Yang, Y.M.; Liu, Y.; Guo, N.; Wang, L.M.; Huang, W.; Wu, L.W.; Sun, D.J.; Gu, W.K. β-catenin mediates fluoride-induced aberrant osteoblasts activity and osteogenesis. Environ. Pollut. 2020, 265, 114734.
- Karner, C.M.; Long, F.X. Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657.
- Rawadi, G.; Roman-Roman, S. Wnt signaling pathway: A new target for the treatment of osteoporosis. Expert Opin. Ther. Targets 2005, 9, 1063–1077.
- Sun, D.J.; Gao, Y.H. Molecular mechanism of pathogenesis of osteofluorosis: A discussion in the view of bony turnover. Chin. J. Endemiol. 2008, 27, 239–241.
- Chen, X.S.; Yu, Y.N.; Yi, W.; Wan, L.B.; Xie, Y. Effect of fluoride on expression of mRNA and protein of Wnt3a and β-catenin in osteoblast of rats. Chin. J. Endemiol. 2013, 32, 140–145.
- Wang, W.P.; Xu, J.; Liu, K.J.; Liu, X.L.; Li, C.C.; Cui, C.Y.; Zhang, Y.Z.; Li, H.B. Suppression of Sclerostin and Dickkopf-1 levels in patients with fluorine bone injury. Environ. Toxicol. Pharmacol. 2013, 35, 402–407.
- Liu, X.L.; Li, C.C.; Liu, K.J.; Cui, C.Y.; Zhang, Y.Z.; Liu, Y. The influence of fluoride on the expression of inhibitors of Wnt/β-catenin signaling pathway in rat skin fibroblast Cells. Biol. Trace Elem. Res. 2012, 148, 117–121.
- Zeng, Q.B.; Xu, Y.Y.; Yu, X.; Yang, J.; Hong, F.; Zhang, A.H. Silencing GSK3β instead of DKK1 can inhibit osteogenic differentiation caused by co-exposure to fluoride and arsenic. Bone 2019, 123, 196–203.
- Yu, J.; Canalis, E. Notch and the regulation of osteoclast differentiation and function. Bone 2020, 138, 115474.
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386.
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos. Int. 2018, 29, 2611–2621.
- Zanotti, S.; Canalis, E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif. Tissue Int. 2012, 90, 69–75.
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 2008, 14, 306–314.
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131.
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253.
- Wang, S.C.; Kawashima, N.; Sakamoto, K.; Katsube, K.I.; Shindo, K.; Suda, H.; Shi, J.N. Osteogenic differentiation of murine mesenchymal progenitor cells Kusa-A1 is promoted by CBF1. Basic Clin. Med. 2006, 26, 409–414.
- Wang, S.C.; Kawashima, N.; Sakamoto, K.; Suda, H.; Shi, J.N. Expression of Notch-related Genes in the Differentiation of Mesenchymal Progenitor Cell, Kusa-A1. J. Oral Sci. Res. 2005, 21, 389–392.
- Chen, X.W.; Wan, C.W.; Xie, C.; Wei, Y.; Wu, Y.; Wan, W. Fluoride Inhibits Expressions of Notch3 and Jag1 Proteins in Rat Bone Tissues. J. Environ. Occup. Med. 2016, 33, 494–498.
- Chen, X.W.; Wan, C.W.; Xie, C.; Yang, X.X.; Wu, Y.; Wan, W. Influence of fluoride on RBPJ and related genes in bone tissue of rats. Chin. J. Public Health 2016, 32, 195–198.
- Zhang, Z.D.; Zhang, X.Z.; Zhao, D.W.; Liu, B.Y.; Wang, B.J.; Yu, W.T.; Li, J.L.; Yu, X.B.; Cao, F.; Zheng, G.S.; et al. TGF-β1 promotes the osteoinduction of human osteoblasts via the PI3K/AKT/mTOR/S6K1 signalling pathway. Mol. Med. Rep. 2019, 19, 3505–3518.
- Ma, J.; Du, D.; Liu, J.; Guo, L.; Li, Y.C.; Chen, A.; Ye, T.W. Hydrogen sulphide promotes osteoclastogenesis by inhibiting autophagy through the PI3K/AKT/mTOR pathway. J. Drug Target. 2020, 28, 176–185.
- Guan, Y.J.; Yang, X.; Yang, W.T.; Charbonneau, C.; Chen, Q. Mechanical activation of mammalian target of rapamycin pathway is required for cartilage development. FASEB J. 2014, 28, 4470–4481.
- Yu, Y.N.; Yang, D.; Zhu, H.Z.; Deng, C.N.; Guan, Z.Z. Expression of mRNA and protein of p38, Osx, PI3K and Akt1 in rat bone with chronic fluorosis. Chin. J. Pathol. 2012, 41, 622–626.
- Chen, R.; Yu, Y.N.; Xu, L.; Deng, C.N. Role of mTOR autophagy signaling in rats cartilages with fluorosis-caused damage. Chin. J. Control Endem. Dis. 2017, 32, 18–19.
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487.
- Zhang, R.X. Fluoride Inhibits the Proliferation and Differentiation of ATDC5 Cells via the PI3K/AKT/mTOR Signaling Pathway. Master’s Thesis, China Medical University, Shenyang, China, 2020.
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog Signaling Pathway Regulates Autophagy in Human Hepatocellular Carcinoma Cells. Hepatology 2013, 58, 995–1010.
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087.
- Long, F.X.; Zhang, X.M.; Karp, S.; Yang, Y.Z.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108.
- Deng, C.N. The Mechanism and Relationship of Hedgehog Signaling in the Bone Injury and Bone Microenvironment of Chronic Fluorosis Rats. Ph.D. Thesis, Guiyang Medical College, Guiyang, China, 2014.
- Gui, C.Z.; Wang, C.S.; Yu, Y.N.; Tang, J.J.; Liu, J.J. Influence of fluoride on the growth and apoptosis of cultured cartilage and antagonizing effect of superoxide dismutase (SOD). Guizhou Med. J. 2004, 28, 291–293.
- Zhang, J.Y.; Xu, S.J.; Wang, K. The Role of Free Radicals in the Pathological Process of Kaschin-Beck Disease Ⅵ. Damage of Type Ⅰ Collagen Induced by Active Oxygen Free Radicals and the Mineralization of Hydroxyapatite in the Damaced Collagen. J. Beijing Med. Univ. 1991, 23, 231–234.
- Fitch, P.M.; Howie, S.E.M.; Wallace, W.A.H. Oxidative damage and TGF-b differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17.
- Zhu, Z.J.; Yu, Y.N.; Chen, R.; Huang, X.L. Role of hedgehog signaling pathway on cartilage tissue damage in chronic fluorosis rats. Chin. J. Public Health 2018, 34, 241–245.
- Datta, N.S.; Abou-Samra, A.B. PTH and PTHrP signaling in osteoblasts. Cell. Signal. 2009, 21, 1245–1254.
- Fermor, B.; Skerry, T.M. PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J. Bone Miner. Res. 1995, 10, 1935–1943.
- Xu, H.; Liu, Q.Y.; Zhang, J.M.; Zhang, H.; Li, G.S. Elevation of PTH and PTHrp induced by excessive fluoride in rats on a calcium-deficient diet. Biol. Trace Elem. Res. 2010, 137, 79–87.
- Yu, X.H.; Yu, H.L.; Jiang, N.N.; Zhang, X.Y.; Zhang, M.M.; Xu, H. PTH (1-34) affects bone turnover governed by osteocytes exposed to fluoride. Toxicol. Lett. 2018, 288, 25–34.
- Neer, R.M.; Arnaud, C.D.; Zanchetta, J.R.; Prince, R.; Gaich, G.A.; Reginster, J.Y.; Hodsman, A.B.; Eriksen, E.F.; Ish-Shalom, S.; Genant, H.K.; et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 2001, 344, 1434–1441.
- Uchida, Y.; Kuroshima, S.; Uto, Y.; Kanai, R.; Inoue, M.; Suzue, M.; Sawase, T. Intermittent administration of parathyroid hormone improves bone quality and quantity around implants in rat tibiae. J. Oral. Biosci. 2020, 62, 139–146.
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806.
- Fulzele, K.; Riddle, R.C.; DiGirolamo, D.J.; Cao, X.M.; Wan, C.; Chen, D.Q.; Faugere, M.C.; Aja, S.; Hussain, M.A.; Brüning, J.C.; et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 2010, 142, 309–319.
- Hu, C.Y.; Ren, L.Q.; Li, X.N.; Wu, N.; Li, G.S.; Liu, Q.Y.; Xu, H. Effect of fluoride on insulin level of rats and insulin receptor expression in the MC3T3-E1 cells. Biol. Trace Elem. Res. 2012, 150, 297–305.
- Yang, C.; Zhang, M.M.; Li, Y.G.; Wang, Y.; Mao, W.X.; Gao, Y.; Xu, H. Streptozotocin Aggravated Osteopathology and Insulin Induced Osteogenesis Through Co-treatment with Fluoride. Biol. Trace Elem. Res. 2015, 168, 453–461.
- Liu, Q.Y.; Liu, H.; Yu, X.H.; Wang, Y.; Yang, C.; Xu, H. Analysis of the Role of Insulin Signaling in Bone Turnover Induced by Fluoride. Biol. Trace Elem. Res. 2016, 171, 380–390.
- Liu, C.; Zhou, X.Y.; Yang, S.R.; Li, Z.W.; Jia, Y. Mechanism and cross-talk of signaling pathways associated with bone damage in fluorosis. J. Environ. Occup. Med. 2021, 38, 794–800.
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844.
- Costa, A.G.; Bilezikian, J.P. Sclerostin: Therapeutic horizons based upon its actions. Curr. Osteoporos. Rep. 2012, 10, 64–72.
- Kramer, I.; Keller, H.; Leupin, O.; Kneissel, M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol. Metab. 2010, 21, 237–244.
- Gui, F.Z. Effects of Fluoride on the Expression of GAG Components and Related Signaling Pathways FGFR3 and Ihh/PTHrP in Rat Growth Plate Cartilage. Master’s Thesis, China Medical College, Shenyang, China, 2019.
- Wang, W.D. Effects of PTH and Notch Signaling Pathway on the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts. Master’s Thesis, Nanjing Medical College, Nanjing, China, 2015.
- Lin, F.T.; Xia, C.; Zhang, B.; Huang, J.G.; Zheng, X.P.; Yi, T.T.; Zhao, H.H.; Zhang, Y.B. Effects of PTHrP and Notch signaling on the proliferation of epiphysis stem cells. Natl. Med. J. China 2011, 91, 2073–2076.
- Day, T.F.; Yang, Y. Wnt and hedgehog signaling pathways in bone development. J. Bone Joint Surg. Am. 2008, 90, 19–24.
- Hu, H.; Hilton, M.J.; Tu, X.; Yu, K.; Ornitz, D.M.; Long, F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 2005, 132, 49–60.
- Ng, R.C.; Matsumaru, D.; Ho, A.S.; Garcia-Barceló, M.M.; Yuan, Z.W.; Smith, D.; Kodjabachian, L.; Tam, P.K.; Yamada, G.; Lui, V.C. Dysregulation of Wnt inhibitory factor 1 (Wif1) expression resulted in aberrant Wnt-β-catenin signaling and cell death of the cloaca endoderm, and anorectal malformations. Cell Death Differ. 2014, 21, 978–989.
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634.
- Surmann-Schmitt, C.; Widmann, N.; Dietz, U.; Saeger, B.; Eitzinger, N.; Nakamura, Y.; Rattel, M.; Latham, R.; Hartmann, C.; von der Mark, H.; et al. Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J. Cell Sci. 2009, 122, 3627–3637.
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205.
- Saidak, Z.; Le Henaff, C.; Azzi, S.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Wnt/β-catenin signaling mediates osteoblast differentiation triggered by peptide-induced α5β1 integrin priming in mesenchymal skeletal cells. J. Biol. Chem. 2015, 290, 6903–6912.
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82.
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells During Fracture Healing. J. Cell Physiol. 2017, 232, 913–921.
- Rivas, A.; Vidal, R.L.; Hetz, C. Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 2015, 19, 1203–1218.
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664.
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183.
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384.
- Shen, X.H.; Zhang, K.Z.; Kaufman, R.J. The unfolded protein response-a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92.
- Xu, H.; Jing, L.; Zhang, C.W.; Qi, L.; Li, G.S. Analysis of proteins in osteoblast exposed to fluoride by two-dimensional electrophoresis and mass spectrometry. Chin. J. Endemiol. 2006, 25, 35–38.
- Maurel, M.; Chevet, E. Endoplasmic reticulum stress signaling: The microRNA connection. Am. J. Physiol. Cell Physiol. 2013, 304, C1117–C1126.
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137.
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91.
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332.
- Zhang, P.F.; Sun, Q.; Zhao, C.Y.; Ling, S.K.; Li, Q.; Chang, Y.Z.; Li, Y.X. HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell. Signal. 2014, 26, 556–563.
- Harding, H.P.; Novoa, I.; Zhang, Y.H.; Zeng, H.Q.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108.
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117.
- Xu, H.; Zhou, Y.L.; Zhang, X.Y.; Lu, P.; Li, G.S. Activation of PERK signaling through fluoride-mediated endoplasmic reticulum stress in OS732 cells. Toxicology 2010, 277, 1–5.
- Sun, F.; Li, X.N.; Yang, C.; Lv, P.; Li, G.S.; Xu, H. A role for PERK in the mechanism underlying fluoride-induced bone turnover. Toxicology 2014, 325, 52–66.
- He, P.; Zhang, M.; He, W.H.; Xia, T.; Yang, K.D.; Wang, A.G. Effects of fluoride on oxidative stress and apoptosis in primary rat hippocampal neurons. Chin. J. Endemiol. 2006, 25, 264–267.
- Shanthakumari, D.; Srinivasalu, S.; Subramanian, S. Effect of fluoride intoxication on lipidperoxidation and antioxidant status in experimental rats. Toxicology 2004, 204, 219–228.
- Xu, H.; Wang, C.H.; Zhao, Z.T.; Zhang, W.B.; Li, G.S. Role of oxidative stress in osteoblasts exposed to sodium fluoride. Biol. Trace Elem. Res. 2008, 123, 109–115.
- Shi, C.L. Protective Effect and Mechanism of Gastrodin on Rats with Chronic Fluorosis and Bone Damage. Master’s Thesis, China Medical University, Shenyang, China, 2019.
- Zhong, Y.F. The Effects of Fluoride on Nrf2-ARE Signal Pathway in Rat Osteoblasts. Master’s Thesis, Guangdong School of Pharmacy, Guangzhou, China, 2011.
- Wang, Z.; Yang, X.; Yang, S.; Ren, G.; Ferreri, M.; Su, Y.; Chen, L.; Han, B. Sodium fluoride suppress proliferation and induce apoptosis through decreased insulin-like growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts. Arch. Toxicol. 2011, 85, 1407–1417.
- Park, K.H.; Park, B.; Yoon, D.S.; Kwon, S.H.; Shin, D.M.; Lee, J.W.; Lee, H.G.; Shim, J.H.; Park, J.H.; Lee, J.M. Zinc inhibits osteoclast differentiation by suppression of Ca2+-Calcineurin-NFATc1 signaling pathway. Cell Commun. Signal. 2013, 11, 74.
- Xie, Y.; Yu, Y.N.; Wan, L.B.; Chen, X.S. Effect of fluoride on expression of CaN mRNA and protein in bone tissue of rats. Chin. J. Pathol. 2012, 41, 761–764.
- Pei, J.R.; Li, B.Y.; Li, Z.W.; Wei, W.; Yao, Y.J.; Xu, J.X.; Gao, Y.H. The effect of fluoride on osteoclast in bone tissue of rats and its mechanism. Chin. J. Endemiol. 2017, 36, 714–718.
- He, H.; Liu, X.; Lv, L.; Liang, H.; Leng, B.; Zhao, D.; Zhang, Y.; Du, Z.; Chen, X.; Li, S.; et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell Death Dis. 2014, 5, e997.
- Neganova, I.; Lako, M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J. Anat. 2008, 213, 30–44.
- Zang, J.J.; Xie, F.; Xu, J.F.; Qin, Y.Y.; Shen, R.X.; Yang, J.M.; He, J. P16 gene hypermethylation and hepatocellular carcinoma: A systematic review and meta-analysis. World J. Gastroenterol. 2011, 17, 3043–3048.
- Chen, C.; Zhang, A.H.; Pan, X.L. The effects of fluoride on hypermethylation, transcription and expression of p16 gene in osteoblasts of rats. Chin. J. Endem. 2016, 35, 89–93.
- Daiwile, A.P.; Tarale, P.; Sivanesan, S.; Naoghare, P.K.; Bafana, A.; Parmar, D.; Kannan, K. Role of fluoride induced epigenetic alterations in the development of skeletal fluorosis. Ecotoxicol. Environ. Saf. 2019, 169, 410–417.
- Lv, H.H.; Tang, X.L.; Fu, S.B. Puerarin reduces methylation of estrogen receptorαpromoter in osteoblasts and regulates its proliferation and osteoblastic differentiation. J. Hebei Med. Univ. 2015, 36, 385–390.
- Zhang, Y.L.; Huang, H.; Gong, B.; Duan, L.Z.; Sun, L.; He, T.K.; Cheng, X.M.; Li, Z.Y.; Cui, L.X.; Ba, Y. Do Environmental Fluoride Exposure and ESRα Genetic Variation Modulate Methylation Modification on Bone Changes in Chinese Farmers? Chem. Res. Toxicol. 2017, 30, 1302–1308.
- Chen, T.; Liu, J. Advances in epigenetic pathogenesis of fluorosis. Chin. J. Endemiol. 2020, 39, 698–702.
- Jiang, Y.T.; Yang, Y.M.; Wang, H.G.; Darko, G.M.; Sun, D.J.; Gao, Y.H. Identification of miR-200c-3p as a major regulator of SaoS2 cells activation induced by fluoride. Chemosphere 2018, 199, 694–701.
- He, S.Y.; Chen, M.; Lin, X.L.; Lv, Z.Q.; Liang, R.Y.; Huang, L.J. Triptolide inhibits PDGF-induced proliferation of ASMCs through G0/G1 cell cycle arrest and suppression of the AKT/NF-κB/cyclinD1 signaling pathway. Eur. J. Pharmacol. 2020, 867, 172811.
- Ouyang, T.; Qin, Y.; Luo, K.K.; Han, X.; Yu, C.; Zhang, A.H.; Pan, X.L. miR-486-3p regulates CyclinD1 and promotes fluoride-induced osteoblast proliferation and activation. Environ. Toxicol. 2021, 36, 1817–1828.
- Gao, J.Y.; Qin, Y.; Luo, K.K.; Wang, X.L.; Yu, C.; Zhang, A.H.; Pan, X.L. Downregulation of miR-4755-5p promotes fluoride-induced osteoblast activation via tageting Cyclin D1. J. Trace. Elem. Med. Biol. 2020, 62, 126626.
- Luo, K.K.; Qin, Y.; Ouyang, T.; Wang, X.L.; Zhang, A.H.; Luo, P.; Pan, X.L. Let-7c-5p Regulates CyclinD1 in Fluoride-Mediated Osteoblast Proliferation and Activation. Toxicol. Sci. 2021, 182, 275–287.
- Kapinas, K.; Kessler, C.; Ricks, T.; Gronowicz, G.; Delany, A.M. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J. Biol. Chem. 2010, 285, 25221–25231.
- Kapinas, K.; Kessler, C.B.; Delany, A.M. miR-29 suppression of osteonectin in osteoblasts: Regulation during differentiation and by canonical Wnt signaling. J. Cell Biochem. 2009, 108, 216–224.
- Wang, T.; Xu, Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem. Biophys. Res. Commun. 2010, 402, 186–189.
- Yang, C.; Wang, Y.; Xu, H. Treatment and Prevention of Skeletal Fluorosis. Biomed. Environ. Sci. 2017, 30, 147–149.
- Wang, W.Y.; Gui, C.Z.; Guan, Z.Z. Research progress on antagonists of fluorosis. Occup. Health 2021, 37, 2433–2438.
- Gupta, S.K.; Gupta, R.C.; Seth, A.K.; Gupta, A. Reversal of fluorosis in children. Acta Paediatr. Jpn. 1996, 38, 513–519.
- Guo, S.Q.; Yu, M.J.; Shen, H.P.; Wang, D.; Yuan, Z.H.; Cheng, J.F. Histomorphometry Effect of Compound Traditional Chinese Medicine on Rats Skeletal Fluorosis. Mod. Prev. Med. 2016, 43, 1471–1475.