Peroxisome proliferator-activated receptors (PPARs) are non-steroid nuclear receptors, which dimerize with the retinoid X receptor (RXR) and bind to PPAR-responsive regulatory elements (PPRE) in the promoter region of target genes. Recently, peroxisome proliferator-activated receptor (PPAR)-α and γ isoforms have been gaining consistent interest in neuropathology and treatment of neuropsychiatric disorders.
- PPAR-α
- PPAR-γ
- neuropsychiatric disorders
- major depression
- Alzheimer’s disease
- allopregnanolone
- BDNF
- neuroinflammation
- toll-like receptor
1. Introduction
2. Brain Distribution of PPARs in the Rodent Brain
3. Neuropsychiatric Disorders and PPARs
3.1. Mood Disorders
Preclinical Studies | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Models of mood disorders | |||||||||||||||||||||||||||||||||
Mood disorders | |||||||||||||||||||||||||||||||||
Disease | |||||||||||||||||||||||||||||||||
MDD | Model | Clinical Trial | Agonist | Rosiglitazone | Molecular Target | PPAR-γ | Effect | Improve symptoms; normalize pro-inflammatory cytokines | References | ||||||||||||||||||||||||
[ | ] | Depression | GW-9662 treatment | ||||||||||||||||||||||||||||||
Double-blind, randomized clinical trial; 24-week. | Rosiglitazone | Pioglitazone | PPAR-γ | PPAR-γ | Antidepressant effect; reduce the immobility time in the forced swim test | Improve anxiety and depression |
[18] |
||||||||||||||||||||||||||
[ | ] |
[57] |
Chronic social defeat stress | WY14643 | PPAR-α | ||||||||||||||||||||||||||||
Bipolar depression | Pioglitazone (15–30 mg/day for 8 weeks) | Improve depressive-like behavior in the tail suspension test and forced swim test | PPAR-γ | [44] |
[ |
Improve depressive symptoms |
36] |
||||||||||||||||||||||||||
[ | ] |
[58] |
Chronic social defeat stress | Fenofibrate | |||||||||||||||||||||||||||||
Double-blind, randomized, placebo-controlled trial | Pioglitazone (15–45 mg/day for 8 weeks) | PPAR-α | PPAR-γ | Antidepressant-like effects |
[45] |
[ |
Fail to improve bipolar depression symptoms37] | ||||||||||||||||||||||||||
[ | ] |
[59] |
CMS-exposed rats | Simvastatin | PPAR-α | Reverse the depression-like behaviors promoting BDNF signaling pathway | |||||||||||||||||||||||||||
Double-blind, randomized, placebo-controlled trial | Pioglitazone (30 mg/day for 12 weeks) | [46] |
[ |
PPAR-γ |
38] |
||||||||||||||||||||||||||||
Differential improvement according to metabolic and depressive status | [ | 68] | CMS-exposed mice | Pioglitazone | PPAR-γ | Decrease microglial activated status (Iba1+) and pro-inflammatory cytokines |
[47] |
[39] |
|||||||||||||||||||||||||
PTSD | Socially isolated mice | Fenofibrate PEA | PPAR-α | Increase brain levels of allopregnanolone; Improve anxiety-like behavior; facilitate contextual fear extinction and fear extinction retention |
[10], |
[41] |
|||||||||||||||||||||||||||
Models of neurodevelopmental disorders | |||||||||||||||||||||||||||||||||
ASD | 16-week prospective study of autistic children | Pioglitazone | PPAR-γ | Improve repetitive and externalizing behaviors, social withdrawal |
[70] |
[62] |
|||||||||||||||||||||||||||
Neurological disorders | Pioglitazone (from postnatal day 24) | PPAR-γ | Mitigate the ASD-like behavior and reduce oxidative stress and inflammation |
[51] |
[43] |
||||||||||||||||||||||||||||
VPA-autism like Wistar rat | Fenofibrate | PPAR-α | Reduce oxidative stress and inflammation in several brain regions |
[52] |
[44] |
||||||||||||||||||||||||||||
BTBR | PEA | PPAR-α | Revert the altered phenotype and improve ASD-like behavior |
[53] |
[45] |
||||||||||||||||||||||||||||
BTBR | GW0742 | PPAR-β/δ | Improve repetitive behaviors and lowers thermal sensitivity responses; decrease pro-inflammatory cytokines |
[54] |
[46] |
||||||||||||||||||||||||||||
Model of neurological disorders | |||||||||||||||||||||||||||||||||
PD | MPTP | Pioglitazone | PPAR-γ | Protect against neurotoxicity; decrease microglial activation and iNOS-positive cells |
[55] |
[47] |
|||||||||||||||||||||||||||
[ | ] | ||||||||||||||||||||||||||||||||
Double-blind, randomized, placebo-controlled trial | Palmitoylethanolamide (PEA) | PPAR-α | Improve depressive symptoms |
[69] |
[61] |
||||||||||||||||||||||||||||
Neurodevelopmental disorders | Schizophrenia | GluN1 knockdown | Pioglitazone | PPAR-γ | Improve long-term memory and help restoring cognitive endophenotypes |
[50] |
[42] |
||||||||||||||||||||||||||
ASD | Propionic acid autism-like rat | ||||||||||||||||||||||||||||||||
AD | Double-blind, randomized, placebo-controlled trial | Pioglitazone (45 mg/day for 18 months) | PPAR-γ | No significant effect |
[71] |
[63] |
|||||||||||||||||||||||||||
MS | Clinical trial, 12 month-treatment | Pioglitazone | PPAR-γ | No improvement in clinical symptoms; decrease grey matter atrophy |
[72] |
[64] |
MPTP | Rosiglitazone | PPAR-γ | Protect from dopaminergic neurons loss; prevents olfactory and motor alteration |
[56] | [49] |
|||||||||||||||||||||
MPTP | MHY908 | PPAR-α/γ dual agonist | Neuroprotective effects; reduce microglial activation and neuroinflammation |
[58] |
[50] |
||||||||||||||||||||||||||||
MPTP | MDG548 | PPAR-γ | Mediate neuroprotection in microglia; promote anti-inflammatory cytokines |
[59] |
[51] |
||||||||||||||||||||||||||||
MPTP | Pioglitazone | PPAR-γ | Decrease microglial activation and iNOS-positive cells |
[60] |
[52] |
||||||||||||||||||||||||||||
Epilepsy | WAG/Rij rats | PEA | PPAR-α | Attenuate seizures |
[61] |
[53] |
|||||||||||||||||||||||||||
AD | Genetically modified AD mouse | Pioglitazone | PPAR-γ | Improve memory and learning deficits; prevent neurodegeneration |
[62] |
[54] |
|||||||||||||||||||||||||||
Streptozotocin rat L165, 041 and F-L-Leu | L165, 041 and F-L-Leu, simultaneously | PPAR-β/δ and PPAR-γ, | Improve myelin and neuronal maturation, mitochondrial proliferation and function; decrease neuroinflammation |
[63] |
[55] |
||||||||||||||||||||||||||||
MS | EAE | Troglitazone | PPAR-γ | Attenuate inflammation |
[64] |
[56] |
|||||||||||||||||||||||||||
Clinical Studies |
|---|
3.2. Neurodevelopmental Disorders
3.3. Neurological Disorders
References
- Chawla, A. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870.
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2016, 54, 2518–2538.
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124.
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications - a review. Nutr. J. 2014, 13, 17.
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, P. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. 2011, 2, 236–240.
- Moreno, S.; Vecchioli, S.F.; Cerù, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neurosci. 2004, 123, 131–145.
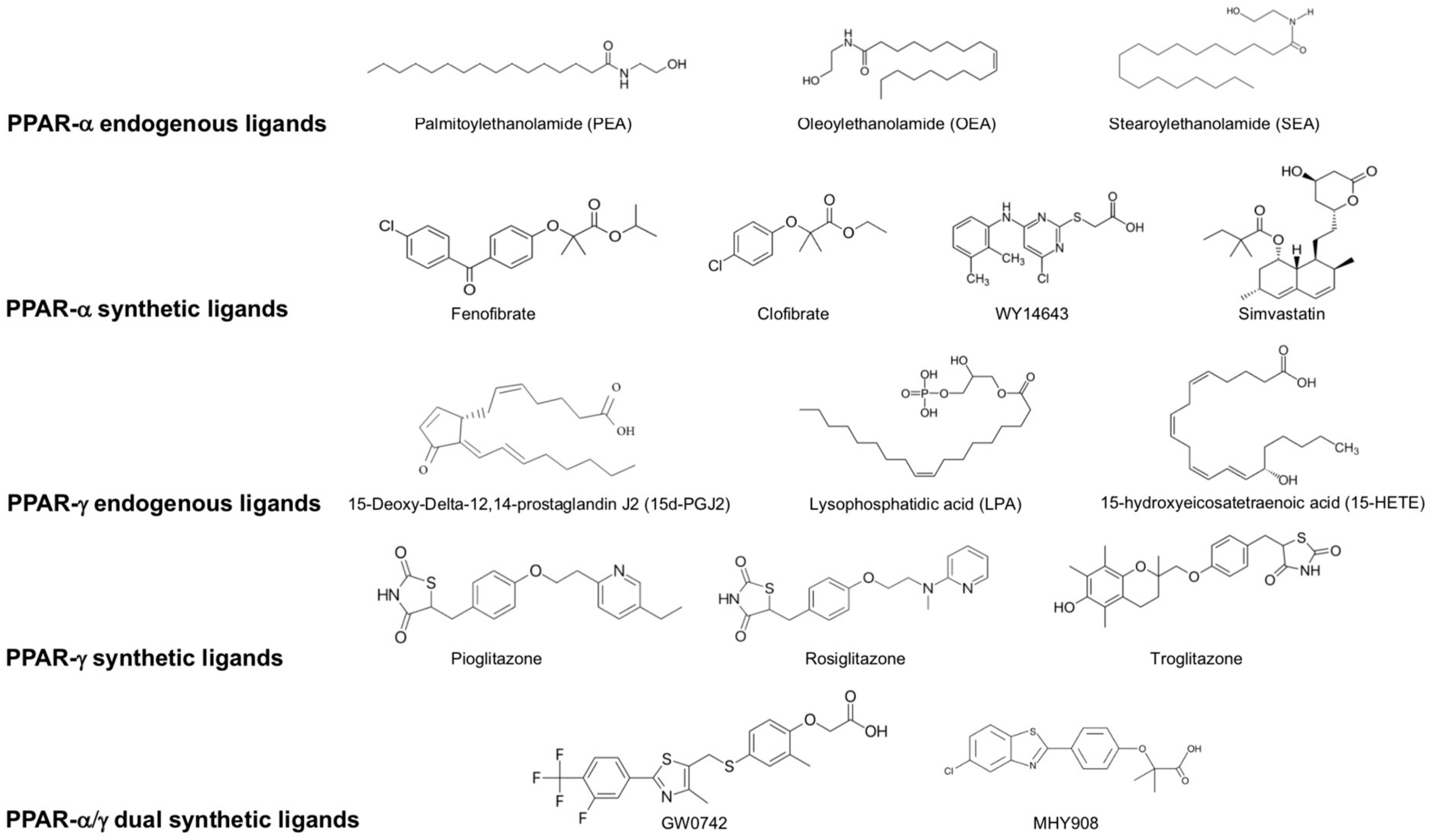
- Nisbett, K.E.; Pinna, G. Emerging therapeutic role of PPAR–α in cognition and emotions. Front. Pharmacol. 2018, 9, 998.
- Rolland, B.; Deguil, J.; Jardri, R.; Cottencin, O.; Thomas, P.; Bordet, R. Therapeutic prospects of PPARs in psychiatric disorders: A comprehensive review. Curr. Drug Targets 2013, 14, 724–732.
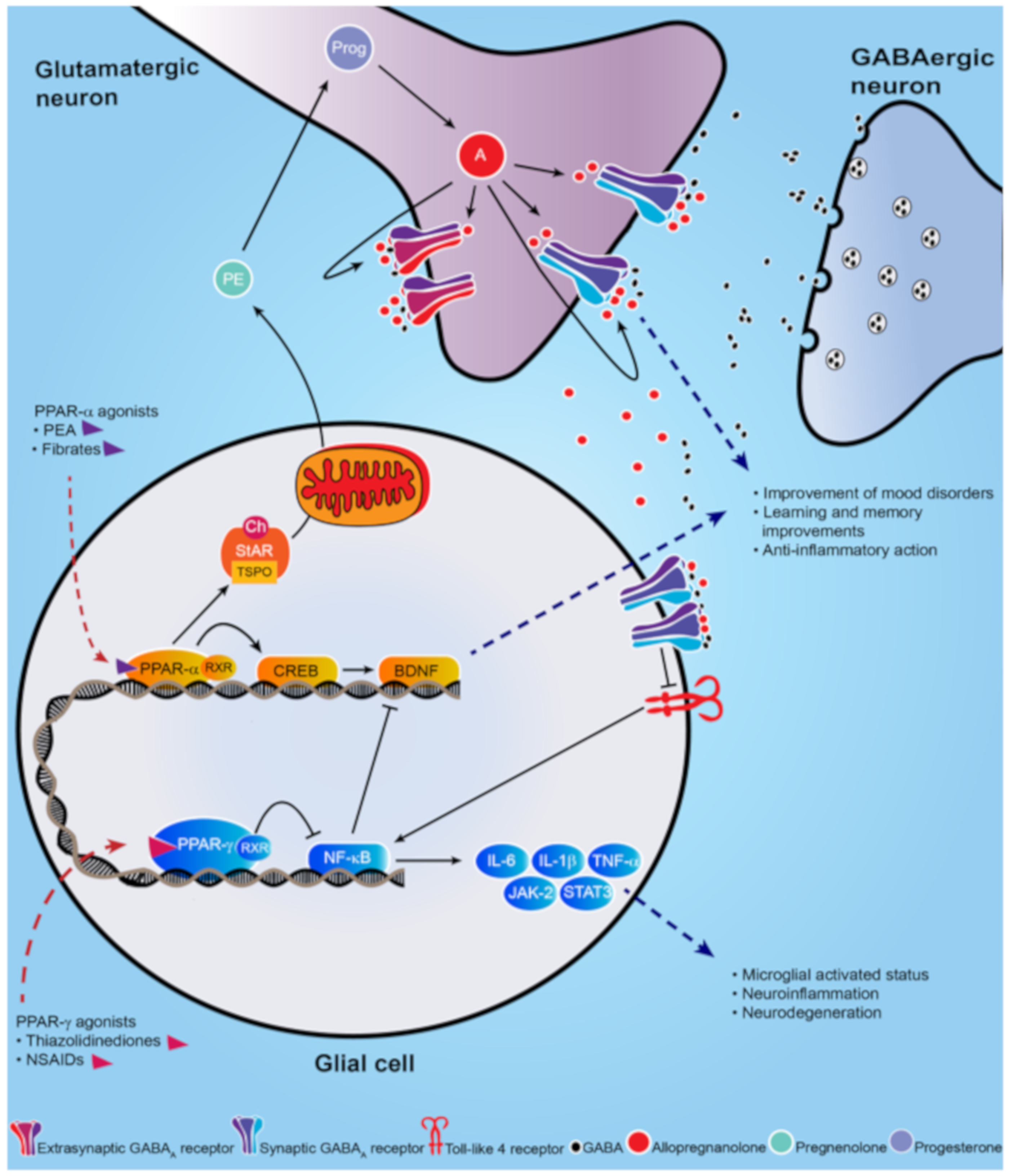
- Locci, A.; Pinna, G. Neurosteroid biosynthesis down-regulation and changes in GABAA receptor subunit composition: a biomarker axis in stress-induced cognitive and emotional impairment. Br. J. Pharmacol. 2017, 174, 3226–3241.
- Locci, A.; Pinna, G. Stimulation of peroxisome proliferator-activated receptor-α by N- palmitoylethanolamine engages allopregnanolone biosynthesis to modulate emotional behavior. Biol. Psychiatry 2019, 85, 1036–1045.
- Balan, I.; Beattie, M.C.; O’Buckley, T.K.; Aurelian, L.; Morrow, A.L. Endogenous neurosteroid (3α,5α)3-hydroxypregnan-20-one inhibits toll-like-4 receptor activation and pro-inflammatory signaling in macrophages and brain. Sci. Rep. 2019, 9, 1220.
- Liao, L.; Zhang, X.; Li, J.; Zhang, Z.; Yang, C.; Rao, C.; Zhou, C.; Zeng, L.; Zhao, L.; Fang, L.; et al. Pioglitazone attenuates lipopolysaccharide-induced depression-like behaviors, modulates NF-κB/IL-6/STAT3, CREB/BDNF pathways and central serotonergic neurotransmission in mice. Int. Immunopharmacol. 2017, 49, 178–186.
- Xu, D.; Lian, D.; Wu, J.; Liu, Y.; Zhu, M.; Sun, J.; He, D.; Li, L. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflammation 2017, 14, 156.
- Lincoff, A.M.; Tardif, J.-C.; Schwartz, G.G.; Nicholls, S.; Rydén, L.; Neal, B.; Malmberg, K.; Wedel, H.; Buse, J.B.; Henry, R.R.; et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus. JAMA 2014, 311, 1515–1525.
- Satirapoj, B.; Watanakijthavonkul, K.; Supasyndh, O. Safety and efficacy of low dose pioglitazone compared with standard dose pioglitazone in type 2 diabetes with chronic kidney disease: A randomized controlled trial. PLoS ONE 2018, 13, e0206722.
- Colle, R.; De Larminat, D.; Rotenberg, S.; Hozer, F.; Hardy, P.; Verstuyft, C.; Feve, B.; Corruble, E. PPAR-? Agonists for the Treatment of Major Depression: A Review. Pharmacopsychiatry 2016, 50, 49–55.
- Ferri, N.; Corsini, A.; Sirtori, C.; Ruscica, M. PPAR-α agonists are still on the rise: An update on clinical and experimental findings. Expert Opin. Investig. Drugs 2017, 26, 593–602.
- Da Rosa, A.; Kaster, M.; Binfaré, R.W.; Morales, S.; Martin-Aparicio, E.; Navarro-Rico, M.L.; Martínez, A.; Medina, M.; Garcia, A.G.; Lopez, M.G.; et al. Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2008, 32, 1549–1556.
- Ahmed, A.A.E.; Al-Rasheed, N.M.; Al-Rasheed, N.M. Antidepressant-like effects of rosiglitazone, a PPARγ agonist, in the rat forced swim and mouse tail suspension tests. Behav. Pharmacol. 2009, 20, 635–642.
- Braissant, O.L.I.V.I.E.R.; Foufelle, F.; Scotto, C.H.R.I.S.T.I.A.N.; Dauça, M.I.C.H.E.L.; Wahli, W.A.L.T.E.R. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366.
- Dickey, A.S.; Pineda, V.V.; Tsunemi, T.; Liu, P.P.; Miranda, H.C.; Gilmore-Hall, S.K.; Lomas, N.; Sampat, K.R.; Buttgereit, A.; Torres, M.-J.M.; et al. PPAR-δ is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat. Med. 2015, 22, 37–45.
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618.
- Aleshin, S.; Strokin, M.; Sergeeva, M.; Reiser, G. nexus of PPAR a - and PPAR c -dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem. Int. 2013, 63, 322–330.
- Scheggi, S.; Melis, M.; De Felice, M.; Aroni, S.; Muntoni, A.L.; Pelliccia, T.; Gambarana, C.; De Montis, M.G.; Pistis, M. PPARα modulation of mesolimbic dopamine transmission rescues depression-related behaviors. Neuropharmacology 2016, 110, 251–259.
- Locci, A.; Pinna, G. Social isolation as a promising animal model of PTSD comorbid suicide: Neurosteroids and cannabinoids as possible treatment options. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2019, 92, 243–259.
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature. 2008, 455, 894–902.
- Dean, J.; Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatry 2017, 27, 101–111.
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, e442.
- Tundo, A.; De Filippis, R.; Proietti, L. Pharmacologic approaches to treatment resistant depression: Evidences and personal experience. World J. Psychiatry 2015, 5, 330–341.
- Albert, P.; Benkelfat, C.; Descarries, L. The neurobiology of depression—revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos. Trans. R. Soc. B: Biol. Sci. 2012, 367, 2378–2381.
- Henter, I.; De Sousa, R.T.; Gold, P.W.; Brunoni, A.R.; Zarate, C.A.; Machado-Vieira, R. Mood Therapeutics: Novel Pharmacological Approaches for Treating Depression. Expert Rev. Clin. Pharmacol. 2017, 10, 153–166.
- Pinna, G.; Rasmusson, A.M. Up-regulation of neurosteroid biosynthesis as a pharmacological strategy to improve behavioural deficits in a putative mouse model of post-traumatic stress disorder. J. Neuroendocr. 2012, 24, 102–116.
- Pinna, G.; Costa, E.; Guidotti, A. Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacol. 2006, 186, 362–372.
- Raber, J.; Arzy, S.; Bertolus, J.B.; DePue, B.E.; Haas, H.E.; Hofmann, S.G.; Kangas, M.; Kensinger, E.; Lowry, C.A.; Marusak, H.A.; et al. Current understanding of fear learning and memory in humans and animal models and the value of a linguistic approach for analyzing fear learning and memory in humans. Neurosci. Biobehav. Rev. 2019, 105, 136–177.
- Rasmusson, A.M.; Marx, C.E.; Pineles, S.L.; Locci, A.; Scioli-Salter, E.R.; Nillni, Y.I.; Liang, J.; Pinna, G. Neuroactive steroids and PTSD treatment. Neurosci. Lett. 2017, 649, 156–163.
- Jiang, B.; Huang, C.; Zhu, Q.; Tong, L.-J.; Zhang, W. WY14643 produces anti-depressant-like effects in mice via the BDNF signaling pathway. Psychopharmacol. 2014, 232, 1629–1642.
- Jiang, B.; Wang, Y.-J.; Wang, H.; Song, L.; Huang, C.; Zhu, Q.; Wu, F.; Zhang, W. Antidepressant-like effects of fenofibrate in mice via the hippocampal brain-derived neurotrophic factor signalling pathway. Br. J. Pharmacol. 2016, 174, 177–194.
- Lin, P.-Y.; Chang, A.Y.; Lin, T.-K. Simvastatin treatment exerts antidepressant-like effect in rats exposed to chronic mild stress. Pharmacol. Biochem. Behav. 2014, 124, 174–179.
- Zhao, Q.; Wu, X.; Yan, S.; Xie, X.; Fan, Y.; Zhang, J.; Peng, C.; You, Z. The antidepressant-like effects of pioglitazone in a chronic mild stress mouse model are associated with PPARγ-mediated alteration of microglial activation phenotypes. J. Neuroinflammation 2016, 13, 259.
- Pibiri, F.; Nelson, M.; Guidotti, A.; Costa, E.; Pinna, G. Decreased corticolimbic allopregnanolone expression during social isolation enhances contextual fear: A model relevant for posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 2008, 105, 5567–5572.
- Pinna, G.; Agís-Balboa, R.C.; Pibiri, F.; Nelson, M.; Guidotti, A.; Costa, E. Neurosteroid Biosynthesis Regulates Sexually Dimorphic Fear and Aggressive Behavior in Mice. Neurochem. Res. 2008, 33, 1990–2007.
- Sullivan, C.R.; Mielnik, C.A.; O’Donovan, S.; Funk, A.J.; Bentea, E.; DePasquale, E.A.; Alganem, K.; Wen, Z.; Haroutunian, V.; Katsel, P.; et al. Connectivity Analyses of Bioenergetic Changes in Schizophrenia: Identification of Novel Treatments. Mol. Neurobiol. 2018, 56, 4492–4517.
- Choi, J.; Lee, S.; Won, J.; Jin, Y.; Hong, Y.; Hur, T.-Y.; Kim, J.-H.; Lee, S.-R.; Hong, Y. Pathophysiological and neurobehavioral characteristics of a propionic acid-mediated autism-like rat model. PLoS ONE 2018, 13, e0192925.
- Mirza, R.; Sharma, B. Benefits of Fenofibrate in prenatal valproic acid-induced autism spectrum disorder related phenotype in rats. Brain Res. Bull. 2019, 147, 36–46.
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Esposito, E.; Calignano, A.; et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: Contribution of central and peripheral mechanisms. Brain Behav. Immun. 2018, 74, 166–175.
- Ahmad, S.F.; Nadeem, A.; Ansari, M.A.; Bakheet, S.A.; Alshammari, M.A.; Attia, S.M. The PPARδ agonist GW0742 restores neuroimmune function by regulating Tim-3 and Th17/Treg-related signaling in the BTBR autistic mouse model. Neurochem. Int. 2018, 120, 251–261.
- Quinn, L.P.; Crook, B.; E Hows, M.; Vidgeon-Hart, M.; Chapman, H.; Upton, N.; Medhurst, A.D.; Virley, D.J. The PPARγ agonist pioglitazone is effective in the MPTP mouse model of Parkinson’s disease through inhibition of monoamine oxidase B. Br. J. Pharmacol. 2008, 154, 226–233.
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with IκBα induction and block of NFκB and iNOS activation. J. Neurochem. 2004, 88, 494–501.
- Schintu, N.; Frau, L.; Ibba, M.; Caboni, P.; Garau, A.; Carboni, E.; Carta, A. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 954–963.
- Cuzzocrea, S. Peroxisome proliferator-activated receptors gamma ligands and ischemia and reperfusion injury. Vasc. Pharmacol. 2004, 41, 187–195.
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARα–leukotriene B4 pathway to inflammation control. Nature. 1996, 384, 39–43.
- Paterniti, I.; Impellizzeri, D.; Crupi, R.; Morabito, R.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Molecular evidence for the involvement of PPAR-δ and PPAR-γ in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J. Neuroinflammation 2013, 10, 20.
- Citraro, R.; Russo, E.; Scicchitano, F.; Van Rijn, C.M.; Cosco, D.; Avagliano, C.; Russo, R.; D’Agostino, G.; Petrosino, S.; Guida, F.; et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacol. 2013, 69, 115–126.
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2016, 26, 97–101.
- Reich, D.; Gallucci, G.; Tong, M.; De La Monte, S.M. Therapeutic Advantages of Dual Targeting of PPAR-δ and PPAR-γ in an Experimental Model of Sporadic Alzheimer’s Disease. J. Park. Dis. Alzheimer’s Dis. 2018, 5, 01–08.
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoé, K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-γ. J. Neuroimmunol. 2001, 116, 40–48.
- Roohafza, H.; Shokouh, P.; Sadeghi, M.; Alikhassy, Z.; Sarrafzadegan, N. A Possible Role for Pioglitazone in the Management of Depressive Symptoms in Metabolic Syndrome Patients (EPICAMP Study): A Double Blind, Randomized Clinical Trial. Int. Sch. Res. Not. 2014, 2014, 1–9.
- Kemp, D.E.; Schinagle, M.; Gao, K.; Conroy, C.; Ganocy, S.J.; Ismail-Beigi, F.; Calabrese, J.R.; Gao, K. PPAR-γ agonism as a modulator of mood: Proof-of-concept for pioglitazone in bipolar depression. CNS Drugs 2014, 28, 571–581.
- Aftab, A.; Kemp, D.E.; Ganocy, S.J.; Schinagle, M.; Conroy, C.; Brownrigg, B.; D’Arcangelo, N.; Goto, T.; Woods, N.; Serrano, M.B.; et al. Double-blind, placebo-controlled trial of pioglitazone for bipolar depression. J. Affect. Disord. 2019, 245, 957–964.
- Lin, K.W.; Wroolie, T.E.; Robakis, T.; Rasgon, N.L. Adjuvant pioglitazone for unremitted depression: Clinical correlates of treatment response. Psychiatry Res. 2015, 230, 846–852.
- Ghazizadeh-Hashemi, M.; Ghajar, A.; Shalbafan, M.-R.; Ghazizadeh-Hashemi, F.; Afarideh, M.; Malekpour, F.; Ghaleiha, A.; Ardebili, M.E.; Akhondzadeh, S. Palmitoylethanolamide as adjunctive therapy in major depressive disorder: A double-blind, randomized and placebo-controlled trial. J. Affect. Disord. 2018, 232, 127–133.
- Capano, L.; Dupuis, A.; Brian, J.; Mankad, D.; Genore, L.; Adams, R.H.; Smile, S.; Lui, T.; Odrobina, D.; Foster, J.A.; et al. A pilot dose finding study of pioglitazone in autistic children. Mol. Autism 2018, 9, 59.
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A Randomized Pilot Clinical Trial of the Safety of Pioglitazone in Treatment of Patients With Alzheimer Disease. Arch. Neurol. 2011, 68, 45–50.
- Kaiser, C.C.; Shukla, D.K.; Stebbins, G.T.; Skias, D.D.; Jeffery, D.R.; Stefoski, D.; Katsamakis, G.; Feinstein, U.L. A pilot test of pioglitazone as an add-on in patients with relapsing remitting multiple sclerosis. J. Neuroimmunol. 2009, 211, 124–130.
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982.
- Martínez-Gras, I.; Perez-Nievas, B.G.; García-Bueno, B.; Madrigal, J.; Andrés-Esteban, E.; Rodríguez-Jiménez, R.; Hoenicka, J.; Palomo, T.; Rubio, G.; Leza, J.C. The anti-inflammatory prostaglandin 15d-PGJ2 and its nuclear receptor PPARgamma are decreased in schizophrenia. Schizophr. Res. 2011, 128, 15–22.
- Chase, K.; Rosen, C.; Gin, H.; Bjorkquist, O.; Feiner, B.; Marvin, R.; Conrin, S.; Sharma, R.P. Metabolic and inflammatory genes in schizophrenia. Psychiatry Res. 2014, 225, 208–211.
- O’Connor, A.M.; Burton, T.J.; Leamey, C.A.; Sawatari, A. The Use of the Puzzle Box as a Means of Assessing the Efficacy of Environmental Enrichment. J. Vis. Exp. 2014, 94, 52225.
- D’Agostino, G.; Cristiano, C.; Lyons, D.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome proliferator-activated receptor alpha plays a crucial role in behavioral repetition and cognitive flexibility in mice. Mol. Metab. 2015, 4, 528–536.
- Magadum, A.; Engel, F.B. PPARβ/δ: Linking Metabolism to Regeneration. Int. J. Mol. Sci. 2018, 19, 2013.
- De Gregorio, D.; Manchia, M.; Carpiniello, B.; Valtorta, F.; Nobile, M.; Gobbi, G.; Comai, S. Role of palmitoylethanolamide (PEA) in depression: Translational evidence. J. Affect. Disord. 2019, 255, 195–200.
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177.
- D’Orio, B.; Fracassi, A.; Ceru, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354.
- Vallée, A.; LeCarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Mol. Neurosci. 2016, 10, 516.
- Papadopoulos, P.; Rosa-Neto, P.; Rochford, J.; Hamel, E. Pioglitazone Improves Reversal Learning and Exerts Mixed Cerebrovascular Effects in a Mouse Model of Alzheimer’s Disease with Combined Amyloid-β and Cerebrovascular Pathology. PLoS ONE 2013, 8, e68612.
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821.
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.N. Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 853–866.