Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Meilian Liu.
Glucocorticoids (GCs), a class of corticosteroids produced by the adrenal cortex in response to stress, exert obesity-promoting effects.
- glucocorticoid
- adiponectin
- WAT
- beige adipocytes
1. Introduction
Brown and beige adipocytes, referred to as lipid-burning cells, dissipate energy in the form of heat, and are widely recognized as a potential therapeutic approach for the treatment of obesity and its associated disorders. The sympathetic nervous system (SNS) richly supports brown adipose tissue (BAT) and is also arborized in white adipose tissue (WAT) at a certain level [1,2][1][2]. Upon cold exposure, SNS releases norepinephrine in adipose tissue, thereby, activating β3 adrenoceptor signaling and thermogenic program in adipocytes [1,2][1][2].
The neuroendocrine hormones, including the thyroid hormones triiodothyronine (T3) and thyroxine (T4), glucocorticoids (GCs), and the adrenal medullary hormones catecholamine and dopamine, play a pivotal role in metabolic homeostasis maintenance [3,4,5][3][4][5] However, the mechanisms underlying neuroendocrine regulation of thermogenic program and cold adaptation remain largely unknown.
GCs are a class of corticosteroids that are critical for the regulation of metabolic, cardiovascular, immunologic, and homeostatic functions in response to various types of stress in humans and in rodents [6,7][6][7]. Although it has been well considered that GCs mobilize glucose, amino acids, and fatty acids, whether GCs are involved in cold-induced thermogenesis remains to be established. Cortisol in humans and corticosterone in rodents are the principal circulating GCs and are secreted under the control of the hypothalamic–pituitary–adrenal (HPA) axis [8]. In mice, fecal corticosterone excretion and plasma adrenocorticotropic hormone (ACTH) increased after 24-hour cold exposure [9].
The correlation between cold stress and the cortisol circulating level has also been assessed in humans, and the effects of cold exposure on serum cortisol levels are contradictory [10,11,12,13,14,15,16][10][11][12][13][14][15][16]. Some studies showed that serum levels of cortisol and/or ACTH were elevated by cold stress [10,11,12,13,14,15][10][11][12][13][14][15]. However, other studies reported a decrease or no effect on the cortisol levels in response to cold exposure [14,16,17][14][16][17]. Therefore, more studies are in need to address the controversy about the correlation between GCs and cold stress.
Stress hormone GCs demonstrate obesogenic and pro-diabetic properties. The increased level of glucocorticoid explains the metabolic abnormalities, such as obesity, hypertension, hyperlipidemia, and glucose intolerance in Cushing’s syndrome with pituitary adenoma [6,18,19][6][18][19]. Chronic GC excess has been also considered to induce obesity at least in part through suppressing BAT activation and browning of WAT in rodents and in humans [20,21,22,23,24,25][20][21][22][23][24][25]. The complexity of GC physiology has been appreciated in the past several years.
Luijten et al. demonstrated that GC administration for 2 weeks lowered UCP1 expression only at thermoneutrality conditions, and the obesity-promoting effects of GC do not require UCP1 under both room temperature and thermoneutrality [26]. Moreover, GC acutely increased BAT activity in humans, implying the primary involvement of GC in cold-induced thermogenesis [27]. In the same study, GC action in human brown adipocytes is distinct from that in rodent cells where cortisol substantially suppressed isoprenaline-stimulated respiration and UCP-1 [27]. While cold-induced fecal corticosterone excretion was reported [9], whether circulating GC senses short-term cold stress and modulates thermogenesis at such condition is largely unknown.
GCs bind to the glucocorticoid receptor (GR) to form a complex and substantially facilitate the translocation of its own receptor into the cell nucleus and transcriptional control of target genes involved in inflammation, gluconeogenesis, and adipocyte differentiation [28,29][28][29]. Upon activation, dimerized GR binds to GC response elements (GREs) and activates gene transcription (Bamberger et al., 1996, Schacke and Rehwinkel, 2004). On the other hand, the Glucocorticoid–GR system has been shown to mediate the promoting effects of chronic stress on adipose tissue inflammation and abnormal glucose and lipid metabolism in obese rats [30].
Constitutive deletion of GR in adipocytes attenuated diet- and aging-induced adiposity accompanied with impaired WAT lipolysis and reduced thermogenesis, whereas had a little effect on BAT UCP1 in short-term cold exposed mice [31]. One recent study showed that conditional adipocyte-specific GR invalidation improved glucose and lipid profiles with an expansion of fat mass [32]. Interestingly, Glantschnig et al. using UCP1-dependent, BAT-specific GR knockout mice demonstrated a negligible role of GR in brown adipocytes in BAT function and systemic metabolism [33]. These studies implied that glucocorticoid-GR signaling plays an unrevealed role in WAT metabolism. However, whether glucocorticoid-GR signaling is required for acute GCs modulation of WAT browning and energy expenditure remains to be established.
2. Circulating Levels of Corticosterone Are Elevated by Acute Cold Stress
Circulating levels of adiponectin were suppressed by cold exposure [42[34][35],43], and this provides a mechanism underlying cold-induced adipose-resident group 2 innate lymphoid cells (ILC2) activation and thermogenesis [35][36]. We asked whether stress-responded neuroendocrine hormones are altered by cold stress.
Similar to norepinephrine levels in WAT [34][37], circulating levels of corticosterone were increased by cold exposure for 48 h in male mice (Figure 1A), while the levels of T3 and adiponectin in serum were decreased by acute cold stress (Figure 1B,C). To investigate whether the downregulation of circulating adiponectin was resulted from the altered neuroendocrine hormones, we treated differentiated 3T3-L1 adipocytes with β3 adrenoceptor agonist CL316,243, T3 and hydrocortisone with different doses for 24 h. We found that the treatment of hydrocortisone but not CL or T3 downregulated both expression levels and secretion levels of adiponectin in adipocytes (Figure 1D), suggesting that acute cold stress downregulates adiponectin expression through GC.
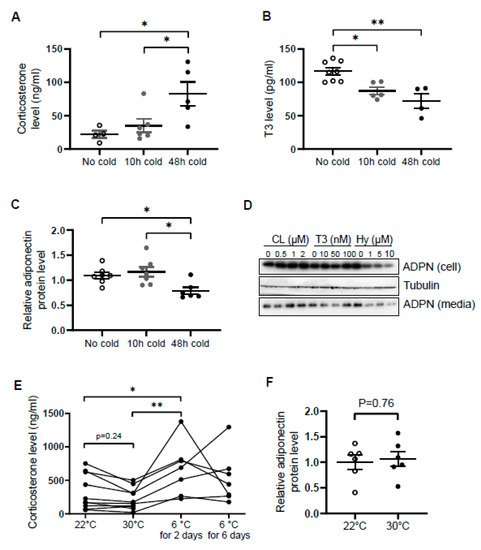
Figure 1. Circulating levels of corticosterone are elevated while adiponectin levels are suppressed by acute cold stress. Acute cold stress increased the circulating levels of corticosterone (A), while suppressed the levels of T3 (B) and adiponectin (C) in serum. White dots, No cold group (mice housed at 22 °C, n = 4–8); gray dots, 10 h cold group (mice housed at 6 °C for 10 h, n = 5–7); black dots, 48 h cold group (mice housed at 6 °C for 48 h, n = 4–6). (D). Treatment of hydrocortisone suppressed expression and release of adiponectin in differentiated 3T3-L1 adipocytes in a dose-dependent manner despite no significant effect when treated with CL or T3 for 24 h. (E) Circulating levels of corticosterone did not significantly alter at 30 °C or after 6 days of cold exposure. Each black dot indicates the individual mouse. (F) Adiponectin expression showed little change between mice housed at 22 °C and 30 °C. White dots, mice housed at 22 °C, n = 6; gray dots, mice housed at 30 °C, n = 6. ADPN, adiponectin. The data in Figure 1A–C are presented as mean ± S.E.M. * p < 0.05, ** p < 0.01.
In agreement with this, the inducing effects of cold stress on GC were diminished during the extension of cold stress (Figure 1E), suggesting an acute elevation of corticosterone by cold stress. In addition, the mild stress from 30 °C to 22 °C had no significant effect on the levels of corticosterone and adiponectin (Figure 1E,F).
3. Hydrocortisone Administration Enhanced Oxygen Consumption and Thermogenic Gene Expression
Elevation of circulating corticosterone by cold stress promoted us to investigate whether corticosterone plays a role in regulating thermogenesis. We administered vehicle or 80 mg/kg hydrocortisone twice a day through intraperitoneal injection for 2 days in 2-month-old male C57/BL6J mice. The results showed that hydrocortisone administration increased oxygen consumption with little effect on food intake, animal activity, and respiratory quotient (RQ) in vivo (Figure 2A). In addition, the expression levels of thermogenic genes Ucp1 and PPARγ in iWAT (Figure 2B) but not in BAT (Figure 2C) were induced by hydrocortisone administration, indicating that corticosterone promotes cold-induced thermogenesis. Consistent with our recent finding that adiponectin inhibits thermogenesis in adipose tissue [35][36], hydrocortisone administration suppresses adiponectin expression in iWAT (Figure 2B) and in circulation (Figure 2D). These results suggest that cold-glucocorticoid pathway may induce thermogenesis by suppressing adiponectin.
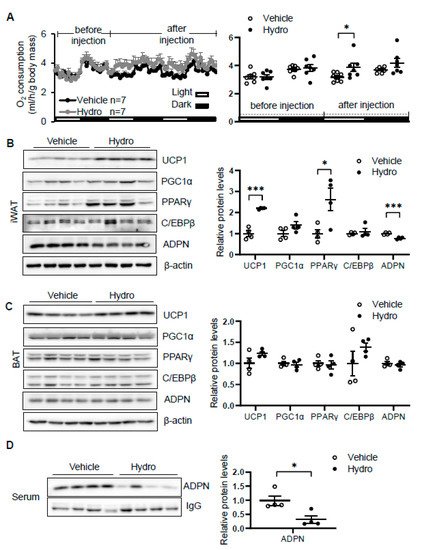
Figure 2. Hydrocortisone increases energy expenditure and suppresses adiponectin expression in adipose tissue. (A) Administration of hydrocortisone increased oxygen (O2) consumption of wild type (WT) mice. The average O2 consumption was normalized to whole-body mass. Hydrocortisone treatment upregulated the expression of thermogenesis genes Ucp1 and Pparγ and downregulated the protein levels of adiponectin in iWAT (B) but not in BAT (C). (D) Hydrocortisone treatment decreased the levels of adiponectin in serum. White dots, vehicle group (n = 4); black dots, hydrocortisone group (n = 4). Hydro, hydrocortisone. The statistical data are presented as the mean ± S.E.M. * p < 0.05, *** p < 0.001.
4. Blocking Glucocorticoid Receptor Signaling Leads to Decreased Energy Expenditure and Thermogenesis in an Adiponectin-Dependent Manner
To dissect the role of glucocorticoid in regulating thermogenesis, we administered mifepristone 5 mg/kg intraperitoneally in 2-month-old male wild type. As a result, administration of mifepristone suppressed cold (6 °C for 48 h)-induced energy expenditure with no effect at room temperature with no significant effects on food intake, animal activities, and respiratory quotient (Figure 3A). Consistent with this, mifepristone treatment downregulated the expression levels of thermogenic genes Ucp1 and Pgc1α in iWAT (Figure 3B) but not in BAT (Figure 2C) under cold condition, supporting the promoting effect of glucocorticoid on thermogenesis. On the other hand, mifepristone treatment induced adiponectin expression in iWAT and in circulation (Figure 3B,D). Furthermore, the suppressing effects of mifepristone on thermogenic gene expression and energy expenditure were abolished in adiponectin deficient mice (Figure 3A,B), suggesting that adiponectin downregulation mediates glucocorticoid-promoted thermogenesis.
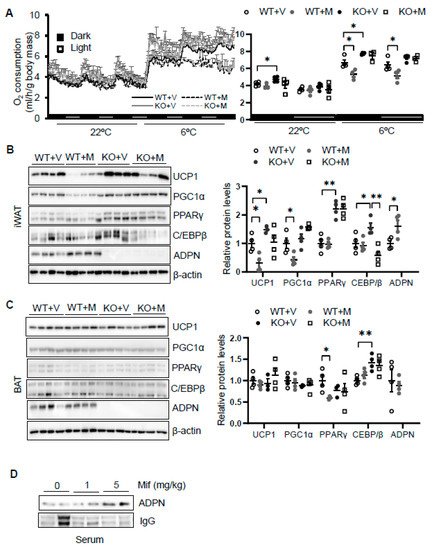
Figure 3. Blocking glucocorticoid receptor signaling inhibits cold-induced energy expenditure through upregulating adiponectin. (A) Administration of mifepristone suppressed acute cold-induced O2 consumption throughout the light and dark cycle. The average O2 consumption was normalized to whole-body mass. WT + V, wild type mice treated with vehicle; WT + M, wild type mice treated with mifepristone; KO+V, adiponectin KO mice treated with vehicle; KO+M, adiponectin KO mice treated with mifepristone. Under cold conditions, the administration of mifepristone downregulated the expression of thermogenesis genes Ucp1 and Pgc1α, and these effects were diminished in iWAT (B) but not in BAT (C) of adiponectin KO mice. White dots, WT mice with vehicle injection (n = 4); gray dots, WT mice with mifepristone injection (n = 5); black dots, adiponectin KO mice with vehicle injection (n = 4); and white square, adiponectin KO mice with mifepristone injection (n = 4). (D) Treatment of mifepristone increased circulating levels of adiponectin. The data in Figure 3A–C are presented as the mean ± S.E.M. * p < 0.05, ** p < 0.01.
5. Hydrocortisone Suppressed Adiponectin Transcription via GR-Dependent Mechanism in Adipocytes
We then asked how glucocorticoid suppresses adiponectin expression. We treated differentiated 3T3-L1 adipocytes with or without hydrocortisone at different doses for 24 h and found that hydrocortisone treatment downregulated adiponectin mRNA and protein levels (Figure 4A and Figure 1D). In contrast, mifepristone treatment upregulated the levels of adiponectin in mRNA and protein as well as the secretion of adiponectin in differentiated 3T3-L1 adipocytes (Figure 4B,C). Along with this, blocking GR with mifepristone rescued the suppressing effect of hydrocortisone on adiponectin expression and secretion (Figure 4D).
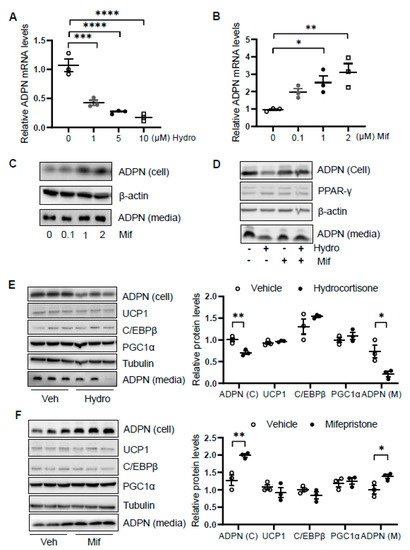
Figure 4. Glucocorticoid receptor signaling transcriptionally suppresses adiponectin in adipocytes. Differentiated 3T3-L1 adipocytes or brown adipocytes were treated with hydrocortisone or mifepristone for 24 h at indicated concentrations. For the co-treatment, 3T3-L1 adipocytes were pre-treated by 2 μM mifepristone for 1 h followed by the co-treatment of 2 μM hydrocortisone. (A) Hydrocortisone treatment suppressed adiponectin mRNA levels in differentiated 3T3-L1 adipocytes in a dose-dependent manner. White dots, vehicle group (n = 3); gray dots, 1 μM hydrocortisone group (n = 3); black dots, 5 μM hydrocortisone group (n = 3); and white square, 10 μM hydrocortisone group (n = 3). Treatment with GC receptor antagonist mifepristone upregulated adiponectin in both mRNA level (B) and protein expression/secretion (C) in a dose-dependent manner in differentiated 3T3-L1 adipocytes. White dots, vehicle group (n = 3); gray dots, 0.1 μM mifepristone group (n = 3); black dots, 1 μM mifepristone group (n = 3); and white square, 2 μM mifepristone group (n = 3). (D) The suppressing effect of hydrocortisone on adiponectin expression and secretion was restored by mifepristone treatment in 3T3-L1 adipocytes. (E) Hydrocortisone treatment suppressed expression and secretion of adiponectin but had no significant effects on the expression of thermogenesis genes in the differentiated brown adipocytes. White dots, vehicle group (n = 3); black dots, hydrocortisone group (n = 3). (F) Mifepristone treatment induced the expression and secretion of adiponectin without significant effects on the expression of thermogenesis genes in the differentiated brown adipocytes. White dots, vehicle group (n = 3); black dots, mifepristone group (n = 3). Veh, vehicle. Hydro, hydrocortisone. Mif, mifepristone. The data in Figure 4A,B,E,F are presented as the mean ± S.E.M. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
In addition, we treated differentiated brown adipocytes and observed that although hydrocortisone treatment suppressed adiponectin expression and secretion, the expression levels of thermogenic genes Ucp1, C/ebpβ, and Pgc1α were little affected in brown adipocytes (Figure 4E). Moreover, mifepristone treatment upregulated adiponectin expression and secretion with little effects on thermogenic gene expression in brown adipocytes (Figure 4F), suggesting that the inducing effect of hydrocortisone on thermogenic gene expression is not mediated by an intrinsic pathway in adipocytes.
6. Glucocorticoid Suppresses Adiponectin Expression by Regulating PPARγ Activity
As the glucocorticoid receptor has been shown to physically interact with PPARγ [7], we investigated if PPARγ is involved in glucocorticoid suppressed adiponectin expression in adipocytes. We found that the suppressing effects of hydrocortisone on adiponectin expression and secretion were reversed by the cotreatment with PPARγ agonist rosiglitazone (Figure 5A,B). Consistent with this, treatment of PPARγ antagonist GW9662 abrogated the inducing effect of mifepristone on adiponectin expression and secretion (Figure 5C,D). These results suggest that glucocorticoid suppresses adiponectin expression in a PPARγ-dependent manner in adipocytes (Figure 5E).
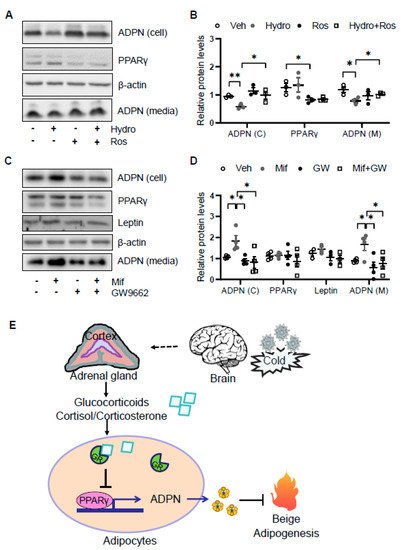
Figure 5. Glucocorticoid receptor signaling suppresses adiponectin expression by inactivating PPARγ. (A) The suppressing effects of GCs on adiponectin expression was attenuated by PPARγ agonist rosiglitazone in differentiated 3T3-L1 adipocytes. Cells were treated with 2 μM rosiglitazone for 1 h followed by the co-treatment of 2 μM hydrocortisone for 24 h. (B) The statistical analysis of the results in Figure 5A. White dots, vehicle group (n = 3); gray dots, hydrocortisone group (n = 3); black dots, rosiglitazone group (n = 3); and white square, hydrocortisone and rosiglitazone cotreatment group (n = 3). (C) The inducing effects of mifepristone on adiponectin were suppressed by the treatment of PPARγ antagonist GW9662 in differentiated 3T3-L1 adipocytes. Cells were treated with 20 μM PPARγ antagonist GW9662 for 1 h followed with the co-treatment of 2 μM mifepristone for 24 h. (D) The statistical analysis of the results in Figure 5C. White dots, vehicle group (n = 4); gray dots, mifepristone group (n = 4); black dots, GW9662 group (n = 4); and white square, mifepristone and GW9662 cotreatment group (n = 4). Hydro, hydrocortisone; Ros, rosiglitazone; Mif, Mifepristone; and GW, GW9662. (E) A schematic model showing that cold stress-elevated GCs promote thermogenesis by suppressing adiponectin production. The data in Figure 5B,D are presented as the mean ± S.E.M. * p < 0.05, ** p < 0.01.
References
- Chi, J.; Wu, Z.; Choi, C.H.J.; Nguyen, L.; Tegegne, S.; Ackerman, S.E.; Crane, A.; Marchildon, F.; Tessier-Lavigne, M.; Cohen, P. Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell Metab. 2018, 27, 226–236.e3.
- Young, J.B.; Saville, E.; Rothwell, N.J.; Stock, M.J.; Landsberg, L. Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. J. Clin. Investig. 1982, 69, 1061–1071.
- Kuliczkowska-Plaksej, J.; Milewicz, A.; Jakubowska, J. Neuroendocrine control of metabolism. Gynecol. Endocrinol. 2012, 28 (Suppl. 1), 27–32.
- Klieverik, L.P.; Coomans, C.P.; Endert, E.; Sauerwein, H.P.; Havekes, L.M.; Voshol, P.J.; Rensen, P.C.; Romijn, J.A.; Kalsbeek, A.; Fliers, E. Thyroid hormone effects on whole-body energy homeostasis and tissue-specific fatty acid uptake in vivo. Endocrinology 2009, 150, 5639–5648.
- Lindmark, S.; Lonn, L.; Wiklund, U.; Tufvesson, M.; Olsson, T.; Eriksson, J.W. Dysregulation of the autonomic nervous system can be a link between visceral adiposity and insulin resistance. Obes. Res. 2005, 13, 717–728.
- Zakrzewska, K.E.; Cusin, I.; Sainsbury, A.; Rohner-Jeanrenaud, F.; Jeanrenaud, B. Glucocorticoids as counterregulatory hormones of leptin: Toward an understanding of leptin resistance. Diabetes 1997, 46, 717–719.
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54.
- Bauer, M.; Caroff, S.; Winokur, A.; Koenig, R. Neuroendocrine responses to cold stress in normal subjects and depressives. Psychoneuroendocrinology 1987, 12, 483–490.
- Van Den Beukel, J.C.; Grefhorst, A.; Quarta, C.; Steenbergen, J.; Mastroberardino, P.G.; Lombes, M.; Delhanty, P.J.; Mazza, R.; Pagotto, U.; Van Der Lely, A.J.; et al. Direct activating effects of adrenocorticotropic hormone (ACTH) on brown adipose tissue are attenuated by corticosterone. FASEB J. 2014, 28, 4857–4867.
- Wilkerson, J.E.; Raven, P.B.; Bolduan, N.W.; Horvath, S.M. Adaptations in man’s adrenal function in response to acute cold stress. J. Appl. Physiol. 1974, 36, 183–189.
- Wagner, J.A.; Horvath, S.M.; Kitagawa, K.; Bolduan, N.W. Comparisons of blood and urinary responses to cold exposures in young and older men and women. J. Gerontol. 1987, 42, 173–179.
- Ohno, H.; Yahata, T.; Yamashita, K.; Kuroshima, A. Effect of acute cold exposure on ACTH and zinc concentrations in human plasma. Jpn. J. Physiol. 1987, 37, 749–755.
- Vigas, M.; Martino, E.; Bukovska, M.; Langer, P. Effect of acute cold exposure and insulin hypoglycemia on plasma thyrotropin levels by IRMA in healthy young males. Endocrinol. Exp. 1988, 22, 229–234.
- Wittert, G.A.; Or, H.K.; Livesey, J.H.; Richards, A.M.; Donald, R.A.; Espiner, E.A. Vasopressin, corticotrophin-releasing factor, and pituitary adrenal responses to acute cold stress in normal humans. J. Clin. Endocrinol. Metab. 1992, 75, 750–755.
- Gerra, G.; Volpi, R.; Delsignore, R.; Maninetti, L.; Caccavari, R.; Vourna, S.; Maestri, D.; Chiodera, P.; Ugolotti, G.; Coiro, V. Sex-related responses of beta-endorphin, ACTH, GH and PRL to cold exposure in humans. Acta. Endocrinol. 1992, 126, 24–28.
- Leppaluoto, J.; Korhonen, I.; Huttunen, P.; Hassi, J. Serum levels of thyroid and adrenal hormones, testosterone, TSH, LH, GH and prolactin in men after a 2-h stay in a cold room. Acta. Physiol. Scand. 1988, 132, 543–548.
- Wilson, O.; Hedner, P.; Laurell, S.; Nosslin, B.; Rerup, C.; Rosengren, E. Thyroid and adrenal response to acute cold exposure in man. J. Appl. Physiol. 1970, 28, 543–548.
- Pasquali, R.; Vicennati, V.; Cacciari, M.; Pagotto, U. The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann. N. Y. Acad. Sci. 2006, 1083, 111–128.
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 2007, 275, 43–61.
- Strack, A.M.; Bradbury, M.J.; Dallman, M.F. Corticosterone decreases nonshivering thermogenesis and increases lipid storage in brown adipose tissue. Am. J. Physiol. 1995, 268, R183–R191.
- Poggioli, R.; Ueta, C.B.; Drigo, R.A.; Castillo, M.; Fonseca, T.L.; Bianco, A.C. Dexamethasone reduces energy expenditure and increases susceptibility to diet-induced obesity in mice. Obesity 2013, 21, E415–E420.
- Van Den Beukel, J.C.; Boon, M.R.; Steenbergen, J.; Rensen, P.C.; Meijer, O.C.; Themmen, A.P.; Grefhorst, A. Cold Exposure Partially Corrects Disturbances in Lipid Metabolism in a Male Mouse Model of Glucocorticoid Excess. Endocrinology 2015, 156, 4115–4128.
- Thuzar, M.; Law, W.P.; Ratnasingam, J.; Jang, C.; Dimeski, G.; Ho, K.K.Y. Glucocorticoids suppress brown adipose tissue function in humans: A double-blind placebo-controlled study. Diabetes Obes. Metab. 2018, 20, 840–848.
- Kong, X.; Yu, J.; Bi, J.; Qi, H.; Di, W.; Wu, L.; Wang, L.; Zha, J.; Lv, S.; Zhang, F.; et al. Glucocorticoids transcriptionally regulate miR-27b expression promoting body fat accumulation via suppressing the browning of WAT. Diabetes 2015, 64, 393–404.
- Lv, Y.F.; Yu, J.; Sheng, Y.L.; Huang, M.; Kong, X.C.; Di, W.J.; Liu, J.; Zhou, H.; Liang, H.; Ding, G.X. Glucocorticoids Suppress the Browning of Adipose Tissue via miR-19b in Male Mice. Endocrinology 2018, 159, 310–322.
- Luijten, I.H.N.; Brooks, K.; Boulet, N.; Shabalina, I.G.; Jaiprakash, A.; Carlsson, B.; Fischer, A.W.; Cannon, B.; Nedergaard, J. Glucocorticoid-Induced Obesity Develops Independently of UCP1. Cell Rep. 2019, 27, 1686–1698.
- Ramage, L.E.; Akyol, M.; Fletcher, A.M.; Forsythe, J.; Nixon, M.; Carter, R.N.; Van Beek, E.J.; Morton, N.M.; Walker, B.R.; Stimson, R.H. Glucocorticoids Acutely Increase Brown Adipose Tissue Activity in Humans, Revealing Species-Specific Differences in UCP-1 Regulation. Cell Metab. 2016, 24, 130–141.
- Bamberger, C.M.; Schulte, H.M.; Chrousos, G.P. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr. Rev. 1996, 17, 245–261.
- Schacke, H.; Rehwinkel, H. Dissociated glucocorticoid receptor ligands. Curr. Opin. Investig. Drugs 2004, 5, 524–528.
- Nagasawa, K.; Matsuura, N.; Takeshita, Y.; Ito, S.; Sano, Y.; Yamada, Y.; Uchinaka, A.; Murohara, T.; Nagata, K. Attenuation of cold stress-induced exacerbation of cardiac and adipose tissue pathology and metabolic disorders in a rat model of metabolic syndrome by the glucocorticoid receptor antagonist RU486. Nutr. Diabetes 2016, 6, e207.
- Mueller, K.M.; Hartmann, K.; Kaltenecker, D.; Vettorazzi, S.; Bauer, M.; Mauser, L.; Amann, S.; Jall, S.; Fischer, K.; Esterbauer, H.; et al. Adipocyte Glucocorticoid Receptor Deficiency Attenuates Aging- and HFD-Induced Obesity and Impairs the Feeding-Fasting Transition. Diabetes 2017, 66, 272–286.
- Dalle, H.; Garcia, M.; Antoine, B.; Boehm, V.; Do, T.T.H.; Buyse, M.; Ledent, T.; Lamaziere, A.; Magnan, C.; Postic, C.; et al. Adipocyte Glucocorticoid Receptor Deficiency Promotes Adipose Tissue Expandability and Improves the Metabolic Profile Under Corticosterone Exposure. Diabetes 2019, 68, 305–317.
- Glantschnig, C.; Mattijssen, F.; Vogl, E.S.; Ali Khan, A.; Rios Garcia, M.; Fischer, K.; Muller, T.; Uhlenhaut, H.; Nawroth, P.; Scheideler, M.; et al. The glucocorticoid receptor in brown adipocytes is dispensable for control of energy homeostasis. Embo. Rep. 2019, 20, e48552.
- Hui, X.; Gu, P.; Zhang, J.; Nie, T.; Pan, Y.; Wu, D.; Feng, T.; Zhong, C.; Wang, Y.; Lam, K.S.; et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab. 2015, 22, 279–290.
- Dong, M.; Yang, X.; Lim, S.; Cao, Z.; Honek, J.; Lu, H.; Zhang, C.; Seki, T.; Hosaka, K.; Wahlberg, E.; et al. Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 2013, 18, 118–129.
- Wang, L.; Luo, Y.; Luo, L.; Wu, D.; Ding, X.; Zheng, H.; Wu, H.; Liu, B.; Yang, X.; Silva, F.; et al. Adiponectin restrains ILC2 activation by AMPK-mediated feedback inhibition of IL-33 signaling. J. Exp. Med. 2021, 218, e20191054110.
- Ding, X.; Luo, Y.; Zhang, X.; Zheng, H.; Yang, X.; Yang, X.; Liu, M. IL-33-driven ILC2/eosinophil axis in fat is induced by sympathetic tone and suppressed by obesity. J. Endocrinol. 2016, 231, 35–48.
More