Together with histone variants and modifications, alterations in nucleosome positioning, non-coding RNAs, and DNA methylation constitute the epigenetic toolkit. DNA methylation describes the chemical modification of the DNA itself by the addition of methyl groups mostly on cytosines, but also on adenines via DNA methyltransferases (DNMTs) [60], with DNMT1 and DNMT3A being the major DNMTs in the CNS [61]. DNA methylation effects, i.a. transcriptional control when occurring at enhancer and promoter sites, alternative promoter choice and alternative splicing [62,63]. At the level of transcriptional regulation, methylated motifs of transcription factor (TF) binding sites physically impede the binding of methyl-sensitive TFs, leading to transcriptional suppression. Furthermore, the interaction of the methyl-CpG-binding domain proteins (MBDs) with methylated DNA prevents binding of TFs and promotes inactive heterochromatin formation by recruiting other chromatin and nucleosome remodeling factors [64].
- Alzheimer
- tauopathy
- TAU
- MAPT
- epigenetics
- neurodegeneration
- neurogenetic disease
- DNA methylation
1. Introduction
2. Implication of DNA Methylation in AD and Tauopathies
2.1. Age-Dependent Changes of DNA Methylation Marks and the Relevance for AD and Tauopathies
2.2. Evidence for the Implication of Altered DNA Methylation Signatures in AD and Tauopathies
Similar to the aging brain, global DNA hypomethylation was reported for AD, supported by decreased immunoreactivity for 5mc in cortical neurons of postmortem AD brains (hippocampus, entorhinal and prefrontal cortex, cerebellum) compared to controls [38][40][41][105,107,108], in line with diminished staining with antibodies directed against DNA methylation maintenance factors in the hippocampal tissue of AD patients [40][107]. Monozygotic twin studies collecting twin pairs discordant for AD found reduced levels of DNA methylation in neuronal nuclei of the AD twin in the temporal neocortex [42][109]. Neuronal and glia cell-type specific differential methylation dynamics associated with AD Braak stage progression were observed for genes such as ANK1, MCF2L, STK32C, LRRC8B, MAP2 and S100B, and methylation changes at the key AD risk genes APP and ADAM17 were identified in a meta-analysis [43][110]. The increased risk of dementia and AD was further correlated with elevated DNA methylation levels in the promoter region of APOE [44][111]. Genetic variation in the APOE gene is related to AD risk and Aβ burden, with the APOE4 variant being the most consistent (see above) genetic risk factor [45][46][112,113]. The DNA methylation-dependent effect was, however, independent of the APOE genotype [44][111]. This points to an independence of allelic and methylation variation of APOE for the risk to develop dementia.2.2.1. DNA Methylation Changes Lead to Pathological Phosphorylation of TAU
Disturbed methylation levels in the promoter regions of genes related to TAU phosphorylation, which plays a critical role in tauopathies, were revealed by diverse clinical and basic research studies in the context of AD [47][114]. GSK3β is the kinase most commonly implicated in hyperphosphorylation of the TAU protein, which in turn is believed to be a prerequisite for the aggregation and formation of NFTs [48][115]. During early AD development, low DNA methylation levels were found in the promoter region of the GSK3β gene (GSK3B) in the prefrontal cortex tissue of AD patients, and consequently GSK3β expression was increased in patients with initial AD [49][116]. While at Braak stages I-II, a decrease of the inactive GSK3β was found in the cortex from AD patients, a considerable increase was observed in AD patients at stages V-VI compared to control subjects. The authors propose that GSK3β hyperactivity, and then NFTs formation, could be initiated at an early stage of the disease and turned off at the final stages [49][116]. TAU hyperphosphorylation is further driven by up-regulated Cdk5 expression, causing diminished long-term synaptic potentiation and culminating in impairments of spatial learning and memory. Low levels of cytosine methylation were detected in the promoter region of Cdk5 in the hippocampal CA1 region in a rat model with Aβ-induced memory deficiency [50][117].2.2.2. Altered DNA Methylation Signatures as a Consequence of Disease Pathophysiology, Such as Aβ Burden and TAU-Phosphorylation
2.2.3. Aβ Peptide and TAU-Phosphorylation-Driven Changes in the Expression and Localization of DNA Repair Related Proteins
2.2.4. Aβ-Associated Changes in DNA Methylation of Cell Cycle-Related Genes
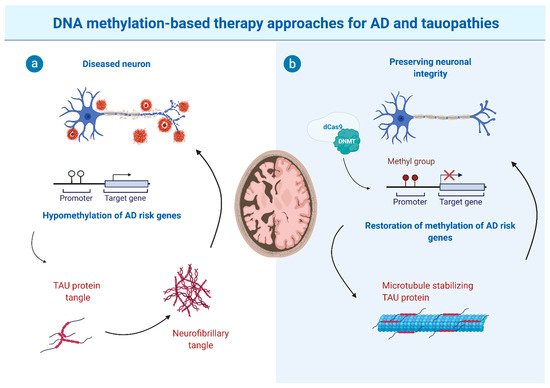
In addition to compromised genomic integrity, dysregulated cell cycle control is an integral part of AD. While in a healthy neuron, abnormal cell cycle reentry leads to apoptosis, abnormal reentry in neurons of aged subjects with AD triggers a cycle of oxidative damage and mitogen production facilitating TAU hyperphosphorylation, Aβ deposition, and CI [60][132]. For genes promoting the activation of cell cycle reentry (i.e., via CDK5), hypomethylation was observed in AD or in AD disease paradigms [61][133]. Exposure of differentiated human neurons to Aβ results in DNA methylation abnormalities of cell-fate genes controlling neuronal differentiation and apoptosis, hinting at a downstream Aβ effect [61][133]. In this context, a recent study described a potential mechanism for DNA methylation-mediated Aβ overproduction, which then triggers Aβ driven hypomethylation of cell cycle-associated genes [62][134]. The same group (Li et al. (2019)) found that AD neurons display significant hypomethylation in the enhancer of the DSCAML1 gene that targets BACE1. BACE1 encodes the β-secretase, which cleaves APP thereby acting on Aβ production. Hence, the DSCAML1 enhancer hypomethylation may activate BACE1 transcription, putatively leading to an increased production of Aβ peptides, resulting in plaques typically preceding the spread of neurofibrillary tangles and neurodegeneration [63][64][135,136]. In agreement with this, changes of DNA methylation signatures in enhancer regulatory elements are frequently observed in AD brains [65][66][137,138]. Together, this indicates that epigenetic impairment of enhancer function is implicated in AD.3. The Potential and Limitations of DNA Methylation-Based Therapy Approaches
As described above, hypomethylation of AD risk genes (such as APP, PSEN1, and PSEN2) was described to be associated with defects in learning and memory. An increase in methyl donor S-adenosyl-L-methionine (SAM) was reported to reduce APP and PSEN1 expression by promoter hypermethylation [67][68][141,142]. In line with this, elevated levels of vitamin B12, folate and other methionine sources in the diet improve methionine bioavailability and were shown to reverse elevated expressions of APP and PSEN1 [69][70][71][143,144,145]. In addition to driving hypermethylation, there is ongoing screening for DNMT inhibitors capable of modulating the methylation of AD or tauopathy risk genes. DNMT inhibitors such as azacitidine and decitabine have already been approved by the FDA for cancer treatment such as leukemia [72][73][74][146,147,148]. The use of DNA demethylating agents has also been used in some other neurodegenerative diseases, such as Friedreich’s ataxia [75][149], which however did not provide promising results in human cells. Finally, due to gene locus-specific changes in DNA methylation signatures, sequence-specific DNA demethylating agents, such as the oligonucleotide antisense inhibitor MG98 [76][77][78][150,151,152], seem promising for future therapeutic approaches to reduce DNA methylation site specifically. Moreover, the hypomethylation of particular genes was described to be implicated in AD and tauopathy pathomechanisms. Hence, locus-specific editing technologies are required for altering or restoring DNA methylation. This can be achieved by clustered regulatory interspaced short palindromic repeats (CRISPR)-deactivated Cas9 (dCas9)-based editing systems that have been described as a specific and efficient method capable of manipulating site-specific DNA methylation [79][153]. This, in combination with improvements in cell type-specific application and blood-brain-barrier overcoming strategies, would open the way for targeted epigenetic therapies (see Figure 1 for schematic depiction).