Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maiko Matsushita and Version 2 by Conner Chen.
During the diagnosis of chronic myelogenous leukemia(CML), patients’ immune cells are derived from leukemic stem cells, and aberrations of both innate and acquired immunity are observed. The following treatment with tyrosine kinase inhibitors (TKIs) not only suppress leukemic cell growth, but also alter immune functions, which may affect prognosis of CML patients.
- chronic myelogenous leukemia
1. Immunogenecity of CML-LSCs
CML-LSCs are enriched in CD34+CD38 low cells [1][2][21,22]. Like other cancer stem cells of solid tumors, LSCs can escape from host immunity by several mechanisms [3][4][23,24]. Tarafdar et al. found that IL-4 gene expression was upregulated in CML-LSCs, which suppresses class II transactivator (CIITA) transcription and subsequently expression of major histocompatibility complex-II (MHC-II) in CML-LSCs compared to normal hematopoietic stem cells (HSCs) [5][25]. The downregulation of MHC-II might reduce antigen presentation against immune cells. Levescot et al. reported that IL-33 binds to the IL-1 receptor ST2 on CML-LSCs and contributes in the maintenance of LSC and imatinib resistance [6][26]. Therefore, CML-LSCs conserve relatively low immunogenicity, and a combination of several therapeutic methods would be required for depleting these cells by immunotherapies.
2. Immune status in CML Patients at Diagnosis
During the diagnosis of CML, patients’ immune cells are derived from CML-LSC, and aberrations of both innate and acquired immunity are observed in these patients [7][8][9][27,28,29]. Hughes et al. investigated the peripheral blood of 21 patients with CML-CP from diagnosis until treatment with TKIs. A decrease in the number of natural killer (NK) cells and cytotoxic T-lymphocytes (CTLs), and impaired responses to leukemia-associated antigens WT1, proteinase 3, BMI-1, and preferentially expressed antigen of melanoma (PRAME) were observed at diagnosis [10][30]. Moreover, the tumor microenvironment of CML cells is immune-inhibitory, characterized by an increase in the number of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) or upregulation of programmed death-1 (PD-1) on CD4/CD8 T cells (Figure 12) [10][11][12][30,31,32]. Immune dysfunction was stronger in high-risk patients [13][33]. In total, anti-CML immune responses were suppressed in patients with CML at diagnosis.
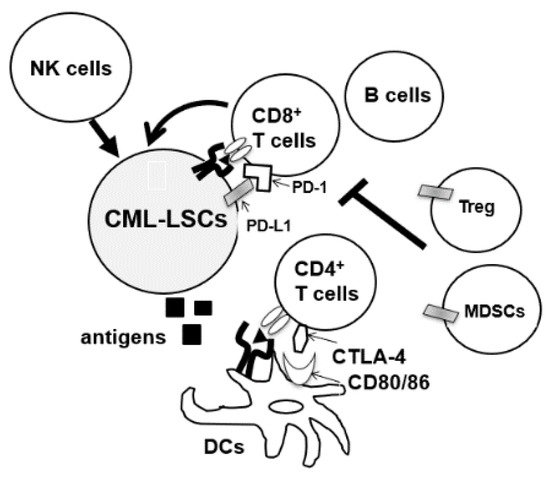
Figure 12. CML-LSCs and immune cells in the tumor microenvironment. NK cells, natural killer cells, MDSCs, myeloid-derived suppressor cells; DCs, dendritic cells, CTLA4, cytotoxic T-lymphocyte-associated protein 4.
3. Immunomodulatory Effects of TKIs in CML
ABL-TKIs not only directly kill CML cells, but also exert immune-modulatory effects. In vitro studies have used peripheral blood mononuclear cells (PBMCs) from healthy donors, showing that TKIs suppress the proliferation of T cells, B cells, and NK cells at therapeutic doses [14][15][16][34,35,36]. However, other clinical studies have shown that TKIs could restore immune inactivated status in CML patients and indirectly suppress CML cells. Since direct killing is less frequently observed in CML-LSCs, these immunomodulatory effects are important for the eradication of CML-LSCs.
3.1. Imatinib
Tanaka et al. have reported that imatinib induces apoptosis of Tregs by its off-target effects on LCK at its therapeutic doses, because imatinib targets a variety of tyrosine kinases other than ABL1, including KIT, PDGFR, LCK, NQO2, and DDR1 [17][37]. They also reported that peripheral blood of imatinib-treated CML patients in complete molecular remission (CMR) had selective depletion of FoxP3hiCD45RA− effector T reg (eTreg) cells, which are in a highly proliferative state and dependent on the TCR signal. They speculated that LCK inhibition attenuates the TCR signal, thereby inducing signal-deprived cell death. This selective depletion of eTregs was not observed in CML patients without CMR. They then stimulated PBMCs of healthy donors with peptides from Melan-A/MART-1, a melanoma-specific tumor antigen, with or without a therapeutic dose of imatinib, and showed that imatinib treatment expanded Melan-A-specific CD8+ T cells accompanied by the depletion of eTregs. An increase in the effector and memory CD8+ T cells and reduction of exhausted CD8+ T cells with PD-1 and LAG-3 expression was observed in CMR patients. Kreutzman et al. showed that the proportion and function of CD56+CD16+NK cells increased withimatinib therapy in CML patients [18][38]. Taken together, the impaired anti-CML immunity can be recovered by imatinib.
3.2. Dasatinib
Dasatinib is known to induce the expansion of large granular lymphocytes (LGLs) [19][39]. Kreutzman et al. reported the development of dasatinib-associated lymphocytosis at an average of three months after the start of treatment and its persistence throughout the therapy. The expanded lymphocytes contained both CD3+CD8+ effector memory T-cells and CD3−CD16+CD56+ NK cells [20][40]. Importantly, the median progression-free survival or overall survival was superior in patients with increased LGL expansion compared to those without [21][41]. Dasatinib is also known to affect Tregs and NK cells. In a prospective phase II clinical trial, D-first study, peripheral blood of 52 patients with newly diagnosed CM-CP was observed for a minimum of 36 months after starting daily administration of 100 mg dasatinib. Treg decreased with dasatinib therapy, and the proportion of Tregs at 12 months of dasatinib treatment was associated with the achievement of deep molecular response (DMR). Treg inhibition was also correlated with an increase in CTL counts, NK cell count, and NK cell differentiation [22][42]. The effects on subsets of NK cells have also been analyzed in several studies. Ishiyama et al. recently reported that CD56− NK cells with PD-1 expression were induced by chronic activation through CMV reactivation in CML or Ph + ALL patients during dasatinib therapy [23][43]. Interestingly, the expansion of CD56− NK cells was associated with a higher rate of DMR, and Wei et al. also reported that dasatinib increased the percentage of Th1 and CD8 T cells and decreased Treg cell levels in the peripheral blood of CML patients who responded to therapy [24][44]. Overall, dasatinib seems to have the potential to enhance both innate and adaptive immunity to eradicate CML-LSCs in some patients. Clinical trials (ClinicalTrials.gov Identifier: NCT04991532) are also ongoing in China to assess the effects of dasatinib on NK cells and T cells of CML-CP patients to compare the immunomodulatory effects of TKIs in clinical settings.
3.3. Nilotinib
In vitro experiments have shown that nilotinib inhibited the proliferation of Tregs; however, it did not induce apoptosis of Tregs at a clinical dose, which is different from imatinib [25][45]. Nilotinib suppresses the differentiation of dendritic cells from peripheral blood monocytes of healthy donors with reduced surface expression of CD83 and CD14. IL-12p70 production from DCs was also impaired after treatment with nilotinib, suggesting that nilotinib has immunosuppressive effects [26][46].
3.4. Bosutinib
Although little is known about the immunomodulatory effects of bosutinib, there is a report analyzing the immune phenotype of newly diagnosed CML patients treated with imatinib (n = 20) or bosutinib (n = 13) [18][38]. No major immunological changes were observed during bosutinib treatment compared to imatinib treatment, possibly because of its narrow spectrum of target kinases.
3.5. Ponatinib
Recently, Leonard et al. reported a combination therapy of blinatumomab and ponatinib for Ph+ ALL. They showed that ponatinib suppressed the expansion of T cells and the production of IFN-γ induced by blinatumomab. They found that this effect was mediated by inhibiting the phosphorylation of LCK. These results suggest that ponatinib might suppress anti-leukemia immunity in CML patients [27][47].
Table 1 summarizes the immunomodulatory effects of TKIs. However, there is little information on the immunomodulatory effects of novel TKIs, including asciminib, which binds to the myristate binding pocket on the kinase domains of ABL1 and BCR-ABL1, and which are effective in patients who are resistant to other TKIs [28][48], and it would be of interest to assess the impact of these drugs on immune cells in the future.
Table 1. Immune-modulatory effects of TKIs.
| TKIs | Immune-Modulatory Effects |
|---|---|
| Imatinib | Treg ↓, effector and memoriy CD8+T cells ↑, exausted T cells ↓, NK cells ↑ |
| Dasatinib | LGL (CD8+T cells or NK cells) ↑,Treg ↓ |
| Nilotinib | Treg ↓DC (differentiation, IL-12 production) ↓ |
| Bosutinib | No change in T cells, monocyte, and granulocytes |
| Ponatinib | CD8+T cells (proliferation IFN-g production) ↓ |
T reg, regulatory T cells; NK cells, natural killer cells; LGL, large granular lymphocytes; DC, dendritic cells. ↑, increase ↓, decrease.