Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Florian Pierre MARTIN.
At the early stages of life development, alveoli are colonized by embryonic macrophages, which become resident alveolar macrophages (ResAM) and self-sustain by local division. Genetic and epigenetic signatures and, to some extent, the functions of ResAM are dictated by the lung microenvironment, which uses cytokines, ligand-receptor interactions, and stroma cells to orchestrate lung homeostasis.
- resident alveolar macrophages
- lung microenvironment
- alveolar niche
- trained immunity
- pneumonia
1. Introduction
Historically, AM were defined by their monocytic origin and high phagocytic functions against pathogens colonizing sterile lungs. The last decades swept these old dogmas away by showing that embryonic macrophages colonize the lung during the first week of life in mice [1] and are in charge of the lung tissue homeostasis [2], where a microbiota naturally dwells [3]. AM play a significant role in pulmonary physiology as some alterations of their functions can lead to severe or even lethal respiratory diseases [4]. Macrophages are also classically separated into two subpopulations: one oriented towards inflammation and antimicrobial defense known as classically activated macrophages (M1), opposing the one oriented towards immune tolerance and tissue repair known as alternatively activated macrophages (M2) [5]. This theoretical dichotomy was recently challenged with in vivo data showing the coexistence of M1 and M2 features in the same macrophages depending on the experimental conditions [6], thus limiting the M1/M2 classification to in vitro models [7].
2. Resident Alveolar Macrophages Ontogeny
Contrary to the former dogma stipulating that all macrophages originated from circulating bone marrow-derived monocytes, ResAM are known for their embryogenic origin, which requires a small contribution from monocytic progenitors during homeostasis. The alveolar colonization by the ResAM occurs during the first week of life in mice, and experiments using irradiated chimeric mice, parabiotic mice, and adoptive cell transfer showed the alveolar colonization was dependent on specific embryonic progenitors [1,8,9][1][8][9]. Yolk sac derived-macrophages, fetal liver monocytes, or bone marrow-derived monocytes can also acquire efficient macrophage functions, such as preventing proteinosis and phagocytosis. Their contribution to the final ResAM pool is not equivalent, the fetal liver monocytes constituting the main proportion of resident macrophages. Many studies that have looked at other tissue-specific macrophages obtained different results, which supposes that every type of macrophage gets its specific preferential embryonic origin. Tissue-specific macrophages from the microglia [10[10][11],11], the liver, the spleen, and the pancreas originate mainly from the yolk sac [12], while intestinal macrophages are constantly replaced by bone marrow-derived monocytes [13]. The contribution from different sources of embryonic progenitors varies in a tissue-dependent manner, and the mechanisms of this tissue imprinting are starting to be elucidated.
3. Maintenance of the Alveolar Macrophage Niche during Homeostasis
The observation that all embryonic macrophage populations (yolk sac-derived, fetal liver-derived, and bone marrow-derived) can independently colonize an empty alveolar niche and differentiate into functional AM suggests that the lung tissue produces maturation and development signals that can turn embryonic progenitors into differentiated AM. In this setting, the production of granulocyte macrophage colony-stimulating factor (GM-CSF) by the lung stroma, specifically by the alveolar epithelial type 2 cells [14], is vital since it induces the expression of the transcription factor peroxisome proliferator-activated receptor gamma (PPARG), a transcription factor essential for the differentiation and perinatal development of AM [15]. The role of GM-CSF is also supported by the demonstration that, in a situation of competition between progenitors, fetal liver progenitors, which have the highest affinity and avidity for this growth factor, become the main contributor to the replenishment of the alveolar niche [8]. As for the differentiation and maintenance of the AM population, the transforming growth factor beta (TGFB) appeared to be crucial in the alveolar niche [16]. The roles and mechanisms of tissue imprinting in AM differentiation and maintenance programs were recently demonstrated. Indeed, while differentiated immune cells usually lose their plasticity, differentiated peritoneal macrophages can acquire the main features of AM after in vivo adoptive transfer into the lungs [17]. This result demonstrated the high plasticity of differentiated macrophages and the role of the tissue microenvironment in both the induction and the maintenance of their functional programming by shaping the chromatin landscape. In summary, even during homeostasis, the maintenance of the functions and identity of AM relies on their local self-proliferation with minimal participation from monocytes, and their constant adaptation to the messages received from the local microenvironment [1,8,9][1][8][9].
4. The Role of Alveolar Macrophages to Maintain Lung Homeostasis
Lungs are not sterile but are continuously exposed to foreign molecules [18] and colonized by a diverse microbiome. The balance between immune response to eliminate these foreign bodies and tolerance is highly regulated in the lungs, with tolerance favored during homeostasis [19,20][19][20]. This is particularly crucial since lung inflammation causes thickening of alveoli walls, jeopardizes gas exchange, and induces life-threatening respiratory failure. Nevertheless, tolerance of virulent pathogens can lead to invasive infections, and it is thus necessary to finely tune the balance between tolerance and immune response.
While the historical theory was that AM induce lung immune tolerance, recent studies have also underlined their central role in maintaining epithelium integrity [21]. AM continuously patrol alveoli to clean the alveolar spaces by phagocytizing inhaled bacteria before they can initiate harmful lung inflammation [22]. AM, which can present antigen but lack co-stimulation molecules, inactivate the CD8+ T cells localized in the airways [23,24][23][24]. In humans, AM closely interact with resident memory T cells, which are central to optimize immune response against inhaled pathogens [25].
All these tolerogenic functions of the AM can be reversed during pneumonia, as we will discuss later, suggesting a microenvironment pressure that remains to be clarified. On the whole-body scale, tissue macrophages differentiation depends on signals given by the macrophage niche [26] but as far as the intrinsic macrophage genetic program is concerned, it is hypothesized that the microenvironment represses proinflammatory genes and promotes an immune tolerance which does make sense in an evolutionary perspective: the new macrophages that replace the dead ones should be set in a tolerogenic mode to prevent unnecessary and potentially deleterious inflammation [27], and this microenvironment pressure applies on AM independently of their ontogeny [17,28,29,30][17][28][29][30] (Figure 1).
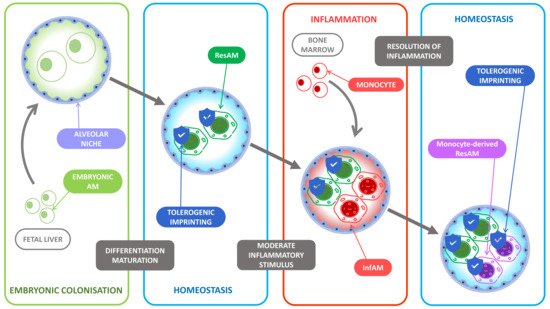
Figure 1. Tolerogenic imprinting throughout inflammation. At birth, the alveolar niche is colonized by fetal liver-derived monocytes. The lung microenvironment then favors their differentiation into resident alveolar macrophages (ResAM) and induces an unalterable tolerogenic imprinting. During inflammation, the main inflammatory actors are the recruited bone marrow-derived monocytes that differentiate into monocyte-derived alveolar macrophages (InfAM) under the pressure of the proinflammatory microenvironment while the ResAM display poor inflammatory functions as most of their tolerogenic imprinting remains. At the resolution of inflammation, the recruited InfAM become monocyte-derived ResAM and undergo the tolerogenic pressure of the microenvironment and display in turn a tolerogenic imprinting to limit further inflammatory response.
Several mediators from the microenvironment participate to the tolerogenic programming of ResAM. First, the metabolic adaptation of ResAM to a local environment poor in nutriments impacts their ability to engulf CD47+ tumor cells and to phagocytize extracellular bodies [31]. The stimulation of CD200R upon the production of CD200 by lung epithelial cells negatively regulates the AM response to bacteria [32]. Epithelial cells, as well as AM themselves, also produce TGFB that limits the activation of AM and promotes their self-maintenance [16,33][16][33]. Another element promoting the tolerance of AM is the surfactant proteins A and D, which are collagen-containing C-type lectins produced by epithelial cells which interact with the signal-regulatory protein alpha (SIRPA) receptor expressed on AM [34]. Surfactant proteins stimulation of SIRPA inhibits the phagocytosis of extracellular bacteria, while the recognition by SIRPA of CD47 precludes the phagocytosis of cells from the host [35,36][35][36]. In summary, the lung microenvironment drives ResAM toward tolerogenic functions, which are critical to prevent potentially detrimental lung inflammation (Figure 2).
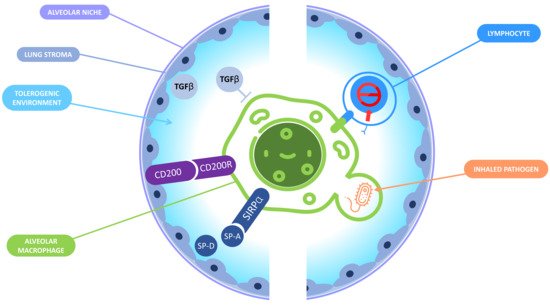
Figure 2. Immune tolerance in the alveolar niche. In the alveolar niche, macrophages and the lung stroma interact together to promote a tolerogenic environment and prevent a deleterious inflammation that could jeopardize the lungs functions. On the one hand, the lung microenvironment induces tolerance in AM: the binding of CD200R expressed by the AM and CD200 expressed by the lung stroma inhibits the macrophages response; lung epithelial cells produce surfactant proteins A and D that can interact with signal-regulatory protein (SIRPA) receptor leading to phagocytosis inhibition; TGB-β is also produced by lung epithelial cells and is responsible for the inactivation of the AM. On the other hand, AM display tolerogenic functions including the inactivation of CD8+ T lymphocytes and the «silent» phagocytosis of inhaled pathogens.
References
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804.
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404.
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963.
- Dranoff, G.; Crawford, A.; Sadelain, M.; Ream, B.; Rashid, A.; Bronson, R.; Dickersin, G.; Bachurski, C.; Mark, E.; Whitsett, J.; et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994, 264, 713–716.
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896.
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288.
- Murray, P.J.; Allen, J.; Biswas, S.K.; Fisher, E.; Gilroy, D.; Goerdt, S.; Gordon, S.; A Hamilton, J.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20.
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992.
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91.
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; de Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb + erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678.
- Tamoutounour, S.; Henri, S.; Lelouard, H.; de Bovis, B.; de Haar, C.; van der Woude, C.J.; Woltman, A.M.; Reyal, Y.; Bonnet, D.; Sichien, D.; et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012, 42, 3150–3166.
- Gschwend, J.; Sherman, S.P.; Ridder, F.; Feng, X.; Liang, H.-E.; Locksley, R.M.; Becher, B.; Schneider, C. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J. Exp. Med. 2021, 218, e20210745.
- Schneider, C.; Nobs, S.P.; O Kurrer, M.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 2014, 15, 1026–1037.
- Yu, X.; Buttgereit, A.; Lelios, I.; Utz, S.G.; Cansever, D.; Becher, B.; Greter, M. The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity 2017, 47, 903–912.e4.
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326.
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Grifffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328.
- Raz, E. Organ-specific regulation of innate immunity. Nat. Immunol. 2007, 8, 3–4.
- Matzinger, P. Friendly and dangerous signals: Is the tissue in control? Nat. Immunol. 2007, 8, 11–13.
- Chikina, A.S.; Nadalin, F.; Maurin, M.; San-Roman, M.; Thomas-Bonafos, T.; Li, X.V.; Lameiras, S.; Baulande, S.; Henri, S.; Malissen, B.; et al. Macrophages maintain epithelium integrity by limiting fungal product absorption. Cell 2020, 183, 411–428.e16.
- Neupane, A.S.; Willson, M.; Chojnacki, A.K.; Castanheira, F.V.E.S.; Morehouse, C.; Carestia, A.; Keller, A.E.; Peiseler, M.; DiGiandomenico, A.; Kelly, M.M.; et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell 2020, 183, 110–125.e11.
- Chelen, C.J.; Fang, Y.; Freeman, G.J.; Secrist, H.; Marshall, J.D.; Hwang, P.T.; Frankel, L.R.; DeKruyff, R.H.; Umetsu, D.T. Human alveolar macrophages present antigen ineffectively due to defective expression of B7 costimulatory cell surface molecules. J. Clin. Investig. 1995, 95, 1415–1421.
- Blumenthal, R.L.; Campbell, D.E.; Hwang, P.; DeKruyff, R.H.; Frankel, L.R.; Umetsu, D.T. Human alveolar macrophages induce functional inactivation in antigen-specific CD4 T cells. J. Allergy Clin. Immunol. 2001, 107, 258–264.
- Snyder, M.E.; Sembrat, J.; Noda, K.; Myerburg, M.M.; Craig, A.; Mitash, N.; Harano, T.; Furukawa, M.; Pilewski, J.; McDyer, J.; et al. Human lung-resident macrophages colocalize with and provide costimulation to PD1hi tissue-resident memory T cells. Am. J. Respir. Crit. Care Med. 2021, 203, 1230–1244.
- Gautier, E.; the Immunological Genome Consortium; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128.
- Guilliams, M.; Svedberg, F.R. Does tissue imprinting restrict macrophage plasticity? Nat. Immunol. 2021, 22, 118–127.
- Gibbings, S.L.; Goyal, R.; Desch, A.N.; Leach, S.M.; Prabagar, M.; Atif, S.M.; Bratton, D.L.; Janssen, W.J.; Jakubzick, C.V. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 2015, 126, 1357–1366.
- Sieweke, M.H. Waddington’s valleys and Captain Cook’s islands. Cell Stem Cell 2015, 16, 7–8.
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460.
- Liu, M.; O’Connor, R.S.; Trefely, S.; Graham, K.; Snyder, N.W.; Beatty, G.L. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated ‘don’t-eat-me’ signal. Nat. Immunol. 2019, 20, 265–275.
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083.
- Mayer, A.K.; Bartz, H.; Fey, F.; Schmidt, L.M.; Dalpke, A.H. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur. J. Immunol. 2008, 38, 1689–1699.
- Janssen, W.J.; McPhillips, K.A.; Dickinson, M.G.; Linderman, D.J.; Morimoto, K.; Xiao, Y.Q.; Oldham, K.M.; Vandivier, R.W.; Henson, P.M.; Gardai, S.J. Surfactant Proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRPα. Am. J. Respir. Crit. Care Med. 2008, 178, 158–167.
- Oldenborg, P.-A.; Gresham, H.D.; Lindberg, F.P. Cd47-signal regulatory protein α (Sirpα) regulates Fcγ and complement receptor–mediated phagocytosis. J. Exp. Med. 2001, 193, 855–862.
- Okazawa, H.; Motegi, S.-I.; Ohyama, N.; Ohnishi, H.; Tomizawa, T.; Kaneko, Y.; Oldenborg, P.-A.; Ishikawa, O.; Matozaki, T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 2005, 174, 2004–2011.
More