Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yara CHAMATA and Version 2 by Vivi Li.
The renin–angiotensin system (RAS) is a key regulator of blood pressure and hypertension. Angiotensin-converting enzyme 2 (ACE2) and angiotensin-converting enzyme I (ACE) are two main components of the RAS that play a major role in blood pressure homeostasis. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses ACE2 as a receptor to enter cells. Despite some controversies, numerous studies have reported a significant association between the use of ACE inhibitors and reduced risk of COVID-19.
- whey peptides
- molecular docking
- hypertension
- ACE2
- COVID-19
- ACE inhibitory activity
- renin–angiotensin system
1. The Counter-Regulatory Renin–Angiotensin System (RAS) Pathways
Among the different regulatory and contra-regulatory systems contributing to the pathogenesis of cardiovascular and renal diseases, the renin angiotensin system (RAS) is one of the main therapeutic targets for CVDs as well as a key player that regulates blood pressure. The RAS pathway includes a cascade of proteases generating some bioactive molecules (Figure 1) [1][5]. Renin is a glycoproteolytic enzyme secreted by the juxtaglomerular cells of the afferent arterioles of the kidney. The liver-derived angiotensinogen acts as the substrate for renin, which cleaves angiotensinogen to form the decapeptide angiotensin I (Ang I) [2][6]. Angiotensin-converting enzyme I (ACE; dipeptidyl carboxypeptidase, EC 3.4.15.1) is a zinc metallopeptidase that is found in male genitals and in vascular endothelial, neuro-epithelial, and absorptive epithelial cells [3][4][5][7,8,9]. It displays both endopeptidase and exopeptidase activities, acting on a wide range of substrates [6][10]. ACE hydrolyzes the inactive decapeptide Ang I into the strong vasoconstrictor angiotensin II (Ang II). Additionally, ACE promotes the inactivation and degradation of the catalytic function of vasodilator bradykinin (BK) into inactive BK (1–7) and BK (1–5) [7][8][9][11,12,13]. By promoting the production of the potent vasoconstrictor Ang II, as well as by degrading the vasodilator BK, ACE plays a dual role in the RAS. In this respect, Ang II is a key component of the RAS pathway, exerting its effects via two G protein-coupled receptors, namely angiotensin type 1 (AT1) and type 2 (AT2). Most of the pathophysiological effects of Ang II are mediated through AT1 receptors, leading to vasoconstriction, cardiovascular inflammation, and aldosterone secretion [10][14]. The AT2 receptor is associated with effects that counteract those of the AT1 receptor; however, many functions of the AT2 receptor are less clear and studies reporting its importance are controversial [11][15].
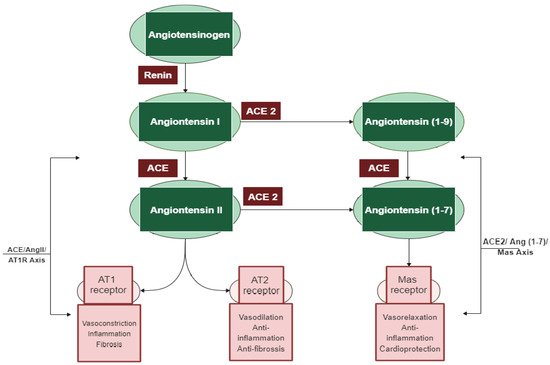
In addition to the classical components of the RAS pathway (renin, ACE, Ang II, AT1, and AT2 receptors), novel peptides such as angiotensin 1–7 (Ang 1–7) and receptors such as angiotensin-converting enzyme 2 (ACE2) appear to play a central role in the regulation of the system [13][16]. ACE2 is an 805 amino-acid Type-I transmembrane protein that functions as a zinc metalloenzyme and monocarboxypeptidase [14][17]. Its extracellular domain consists of a single catalytic metallopeptidase that shares 61% sequence similarity and 42% sequence identity with the catalytic domain of ACE. ACE2 is active and expressed in most tissues; however, the highest expression of ACE2 is mainly observed in vascular endothelial cells and in the renal tubular epithelium [15][18]. Ang II appears to be the major substrate for ACE2 [15][16][18,19]. Ang (1–7) is a vasodilator with anti-proliferative effects produced by the catalytic activity of ACE and ACE2 from Ang I or Ang II [13][16]. The biologically active peptide Ang (1–9) is formed through the hydrolysis of the amino acid leucine from the C-terminal of Ang I by ACE2 (Figure 1) [15][18]. Ang (1–9) is subsequently cleaved by ACE and the neutral endopeptidase 24.11 (NEP) to generate Ang (1–7) [16][19], which can also be generated directly through the cleavage of the amino acid phenylalanine at the C-terminal of Ang II [17][20]. ACE2 therefore plays a key role as a regulator of the RAS pathway through degrading the vasoconstrictor/proliferative peptide Ang II and producing the vasodilator/antiproliferative peptide Ang (1–7) [9][13]. Additionally, the identification of the G protein-coupled receptor Mas, as a receptor of Ang (1–7), was another pivotal step to establish the relevance of Ang (1–7) [18][21]. The ACE2/Ang (1–7)/Mas axis is now accepted to counteract most of the harmful actions of the ACE/Ang II/AT1 receptor axis [19][22]. ACE2 is thus a key counter regulatory enzyme and a potent modulator of blood pressure [12][23].
The discovery of these novel components (ACE2, Ang (1–7), and Mas receptor), which have been recently added to the RAS system, has completely altered our understanding of the regulatory mechanisms of this pathway. It is now widely accepted that the system is dual and consists of two axes: the primarily deleterious axis consisting of ACE/Ang II/AT1 and the beneficial axis consisting of ACE2/Ang-(1–7)/Mas. These novel RAS elements thus open up new opportunities for interfering with the activity of the system and invigorating the development of new cardiovascular drugs targeting the beneficial and counter-regulatory axis of the RAS [20][24].
2. Controversies Regarding the Role of ACE2 in COVID-19
Besides its role as a SARS-CoV-2 receptor, ACE2 is established for its role in hypertension by negatively regulating the RAS through modulating blood pressure to maintain blood pressure homeostasis. The unique interaction of SARS-CoV-2 and host cell receptor ACE2 provides a critical link between COVID-19, hypertension, and CVD [21][22][37,38]. As ACE2 has been identified as the crucial receptor for SARS-CoV-2, the entire RAS should be evaluated when addressing the COVID-19 pandemic. According to Sriram and Insel [23][39], the imbalance in the action of ACE2 and ACE is one of the main culprits of COVID-19 pathobiology. Hence, in order to treat ACE-2-mediated COVID-19, there are two primary approaches suggested to restore ACE/ACE2 balance in the literature: (i) ACE inhibitors/increasing ACE2 or Ang (1–7) levels, and/or (ii) seeking new molecules targeting the S protein or ACE2 receptor to prevent infection by SARS-CoV-2 [24][25][26][40,41,42].
From a therapeutic perspective, activating the ACE2/Ang (1–7)/Mas axis or inhibiting the ACE/Ang II/AT1R axis could be promising [27][28][29][36,43,44]. Previous research has demonstrated that SARS-CoV infection significantly decreases ACE2 levels in infected mice [30][45]. The binding of SARS-CoV-2 to ACE2 also reduces levels of ACE2, thereby inhibiting the ACE2/Ang (1–7) pathway and shifting the balance of the RAS system, consequently leading to the exacerbation of acute severe pneumonia [31][46]. By inhibiting the conversion of Ang I to Ang II, ACE inhibitors reduce Ang II production and subsequently the effects are triggered by its interaction with the receptor AT1R, namely vasoconstriction [12][23]. The hypothetical association between treatment with ACE inhibitors and severe COVID-19 disease has been intensely debated in the literature [32][33][34][35][47,48,49,50]. One hypothesis suggests that the use of these drugs could be harmful in the sense that increased expression of ACE2 receptors may enhance viral-binding and entry [36][37][51,52]. The other hypothesis proposes that ACE inhibitors could be protective by decreasing the production of Ang II and boosting the production of Ang (1–7), which attenuates inflammation and fibrosis, and hence could attenuate lung injury [38][39][53,54]. Various large cohort studies have suggested that the use of ACE inhibitors was not correlated with increased SARS-CoV-2 infection but was in fact linked to a reduced risk of mortality in hospitalized COVID-19 patients [40][41][42][55,56,57].
3. Whey-Derived Peptides as Promising Therapeutic Candidates
Ever since the pandemic brought chaos to lives across the globe, scientists have been making extraordinary efforts to explore therapeutic candidates, such as developing effective vaccines and drugs against COVID-19, to reduce the severity of the outbreak. Given the significance of the ACE2 receptor, research groups have been seeking new molecules targeting this receptor to prevent infection by SARS-CoV-2 and mitigate the development of COVID-19 disease [22][41][43][38,56,58]. Many of the recent studies have investigated the potential of chemical compounds such as flavonols [44][59] and peptides as novel therapeutic inhibitors against SARS-CoV-2, targeting the ACE2 receptor [24][25][26][40,41,42]. Peptide and peptide-based inhibitors represent attractive candidates that hold great promise for the development of ACE2 inhibitors due to their safety, specificity, and efficacy compared to small molecule drugs. Antiviral peptides can also be rationally designed and optimized based on the known structures of viral proteins, as these can be developed to be highly specific for their respective targets [45][46][60,61]. Strategies to interfere with the interaction of the S protein with the ACE2 receptor have been previously examined with SARS-CoV [47][62]. Hence, antagonist ACE2 proteins or their derived peptides may not only be a treatment for preventing the spread of SARS-CoV-2 but also for the modulation of the RAS. These proteins and the derived peptides could be used both to protect patients with COVID-19 disease and to limit the spread of the current SARS-CoV-2 and other coronaviruses by preventing the replication of the virus and development of SARS in the lung [46][61].
Computational approaches play a considerable role in the process of rapid drug development and discovery in a cost and time-efficient manner. In the literature, many researchers have aimed to identify novel ACE2 inhibitors utilizing a molecular docking strategy [24][25][26][40,41,42].
Given the high sequence similarity and sequence identity between ACE and ACE2 [15][18], and the reported reduced risk of mortality and disease associated with use of ACE inhibitors among COVID-19 patients [40][41][42][55,56,57], investigating the ability of ACE inhibitors to block ACE2 interaction with the SARS-CoV-2 S protein would be promising. Various animal and plant proteins have been used in the development of functional foods providing ACE inhibitory activity; however, milk is the main source of antihypertensive ACE-inhibitory peptides reported to date [48][49][63,64]. In fact, previous studies have demonstrated that milk-derived peptides are associated with lower blood pressure [50][65], with some researchers generating evidence to support the beneficial impact of milk proteins on vascular health [51][66].
In our previous work, we characterized ACE inhibitory peptides produced by enzymatic hydrolysis of whey proteins [52][67]. Peptide sequences were identified as major peptides in fractions from the enzymatic hydrolysates CDP (casein-derived peptides) and β-lactoglobulin. The well-known antihypertensive peptide Ile-Pro-Pro (IPP), along with some other novel peptide sequences that have structural similarities with the reported ACE inhibitory peptides, such as Leu-Val-Tyr-Pro-Phe-Pro (LVYPFP), Leu-Ile-Val-Thr-Gln (LIVTQ), and Ile-Ile-Ala-Glu (IIAE), was characterized and identified by a combination of chemical characterization (LC/MS; MS/MS) and structure-activity relationship data. These peptides produced naturally from whey by enzymatic hydrolysis interacted with residues of human ACE, in common with potent ACE-inhibitory drugs, such as Sampatrilat, Captopril, Lisinopril, and Elanapril, which suggests that these natural peptides may be potent ACE inhibitors [52][53][67,68]. The present study aims to explore the efficacy of a natural therapeutic strategy that targets both RAS axes for potential treatment and/or prevention of COVID-19. Herein, we investigate the potential interactions between whey protein-derived peptides with high ACE inhibitory activity (IPP, IIAE, LIVTQ, and LVYPFP) and human ACE2, employing a molecular docking approach. The overall aim is to obtain an improved understanding of such peptides’ function as RAS inhibitors and to assess their potential therapeutic role at the heart of the COVID-19 pandemic.
4. Molecular Docking
In this study, molecular docking was conducted to elicit the potential molecular interactions between the specific amino acids at the binding site of human ACE2 and our previously identified whey protein-derived peptide sequences with high ACE inhibitory activities.
The peptide sequences were docked into the binding site of human ACE2 using the X-ray crystallographic structure of the human ACE2 receptor (PDB code 6M0J). As 6M0J does not contain a co-crystallized ligand, to validate our docking approach, we used the co-crystallized MLN-4760 ACE2 receptor complex (PDB code 1R4L), whereby we extracted the co-crystallized ligand MLN-4760 and re-docked it into the prepared protein 1R4L. The root-mean square deviation (RMSD) between the docked conformation (as generated by superimposition in the program PyMol) and the native co-crystallized ligand conformation was 0.3 Å, which is well within the 2 Å grid spacing used in the docking procedure, demonstrating that the docking method used was valid and reliable. Furthermore, the interactions between the docked ligand and the prepared target receptor mimicked those observed in the crystal structure of the same protein (PDB code 1R4L) [54][69].
To further validate our method, the ligand Carnosine was docked into the prepared X-ray crystal structure of the human ACE2 receptor (PDB code 6M0J) to be used for subsequent docking runs. As Q9BYF1 is the UniPROT code for both 6M0J and 2AJF structures and given that these are 100% identical ACE2 sequences in both X-ray crystal structures (Figures S1–S3), according to the EMBOSS needle results (Figure S4), the interactions between the docked ligand Carnosine and those observed in the crystal structure (PDB code 2AJF) were compared (Figure 2a and Figure S5) [24][40]. In our docking study, Carnosine interacted with key amino acid residues, namely Glu 375, His 378, Glu 402, and Tyr 515 in the active site of ACE2, in accordance with observations reported in the literature [24][54][55][56][40,69,70,71]. In a study providing structural insights for the differences in the inhibition pattern and substrate specificity for ACE and ACE2, amino acid residues His 374, His 378, Glu 375, Tyr 515, Glu 402, and Glu 406 were characterized in the active site of ACE2 [55][70]. These observations were corroborated in the first reported crystal structure of ACE2 in its native and inhibitor-bound states, where key binding residues His 374, His 378, and Glu 402 were identified [54][69]. According to Saadah et al. [24][40], Carnosine interacted with amino acid residues His 378, Glu 402, and Tyr 515 at the active site of ACE2, which was also confirmed in our docking approach.
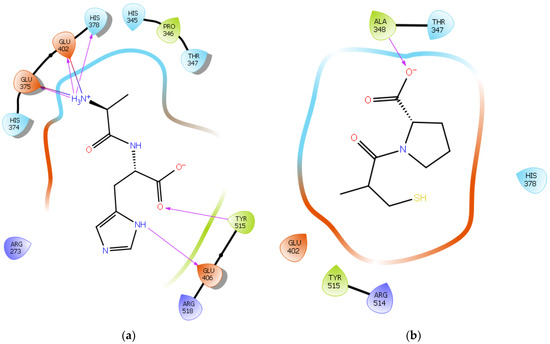
Figure 2. (a) Docking results of the peptide Carnosine in the human angiotensin-converting enzyme 2 (ACE2) active site. The interactions of human ACE2 residues with Carnosine (represented in black) are indicated by arrows of different colors, with purple representing hydrogen bond interactions and blue arrows representing salt bridge interactions. (b) Docking results of the synthetic drug Captopril in the human angiotensin-converting enzyme 2 (ACE2) active site. The interaction of human ACE2 residues with Captopril (represented in black) is indicated by a purple arrow representing hydrogen bond interactions.
The synthetic ACE inhibitory drug Captopril was also docked into 6M0J. According to the docking results, Captopril did not interact with any key binding residues in the active site of ACE2 and formed only one potential hydrogen bond with the backbone of the amino acid residue Ala 348 (Figure 2b and Figure S6). These observations are in line with those reported in other studies, wherein ACE inhibitors such as Captopril could not inhibit ACE2 [55][57][70,72].
Following validation, the human ACE2 receptor (PDB code 6M0J) was then used as the target molecule for docking the peptide sequences of interest into its active site.
Hydrogen bond interactions play a crucial role in the specificity and stability of protein–ligand interactions. The results of ligand-driven docking into the binding site of ACE2 are summarized in Figure 3, Figure 4, Figure 5 and Figure 6, and Figures S7–S10, and Table 1 and Table S1. It is known that His 374, His 378, and Glu 402 are important ligand-binding residues in the zinc-binding site of ACE2 [54][55][56][57][69,70,71,72]. In the current study, IPP showed potential interactions with the key residues His 378 and Glu 402 through hydrogen bond interactions at distances of 2.4 Å and 2.9 Å, respectively. Interestingly, IPP also interacted with these two amino acid residues, namely His 378 and Glu 402, similarly to Carnosine, the best-known drug candidate to match an ACE2 inhibitor structure [24][40]. Additionally, IPP formed a salt bridge and a hydrogen bond with amino acid residue Glu 375, another key active amino acid residue in ACE2 (Table 1, Figure 3) [55][56][70,71]. IIAE, LIVTQ, and LVYPFP also interacted with residue Glu 402 in common with the potent ACE2 inhibitor Carnosine. This was done via hydrogen bonding and salt bridge interaction at distances of 2.9 Å and 4.3 Å, respectively, for IIAE (Table 1, Figure 4); through two hydrogen bonds at distances of 2 Å and 2.6 Å, and through one salt bridge interaction at a distance of 4.3 Å for LIVTQ (Table 1, Figure 5); and through hydrogen bonding and salt bridge interactions at distances of 2.9 and 3.8 Å, respectively, for LVYPFP (Table 1, Figure 6). Additionally, IIAE formed two hydrogen bonds and one salt bridge interaction with key binding amino acid residue Glu 375 (Table 1, Figure 4) [55][56][70,71].
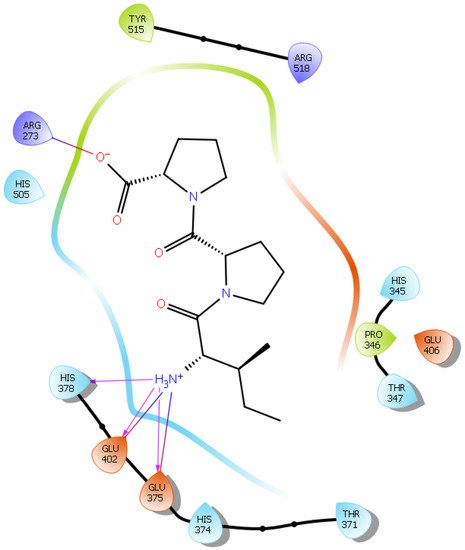
Figure 3. Docking results of the peptide IPP in the active site of the human angiotensin 2-converting enzyme (ACE2). Interactions of human ACE2 residues with the peptide IPP (represented in black) are indicated by arrows of different colors, with purple arrows representing hydrogen bond interactions and blue arrows representing salt bridge interactions.
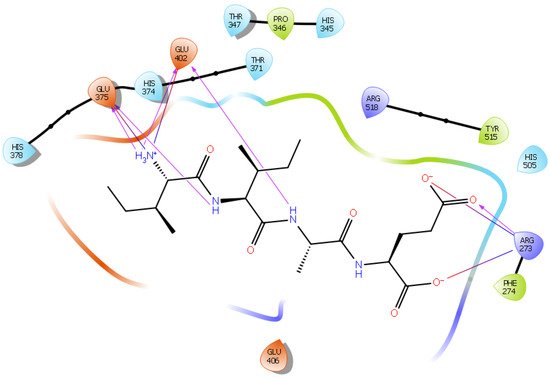
Figure 4. Docking results of the peptide IIAE in the active site of human ACE2. The interactions of human ACE2 residues with the peptide IIAE (represented in black) are indicated by arrows of different colors, with purple arrows representing hydrogen bond interactions and blue arrows representing salt bridge interactions.
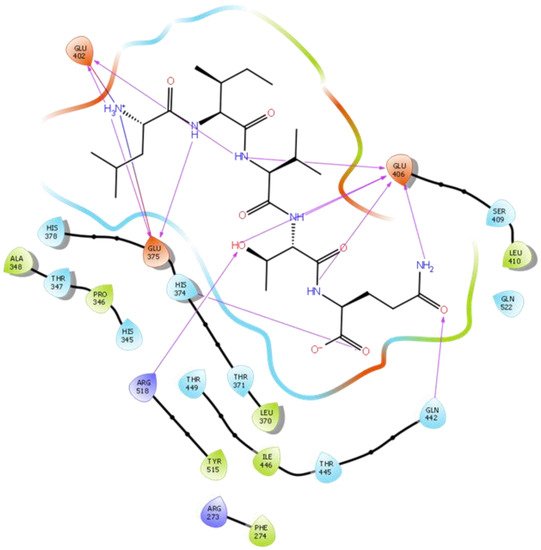
Figure 5. Docking results of the peptide LIVTQ in the human ACE2 active site. The interactions of human ACE2 residues with the peptide LIVTQ (represented in black) are indicated by arrows of different colors, with purple arrows representing hydrogen bond interactions and blue arrows representing salt bridge interactions.
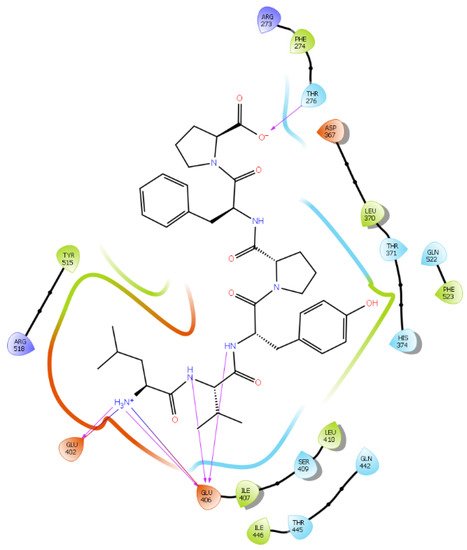
Figure 6. Docking results of the peptide LVYPFP in the human ACE2 active site. The interactions of human ACE2 residues with the peptide (represented in black) are indicated by arrows of different colors, with purple arrows representing hydrogen bond interactions and blue arrows representing salt bridge interactions.
Table 1. Docking results of IPP, IIAE, LIVTQ, and LVYPFP.
| Protein 6MOJ | Ligand IPP | ||
|---|---|---|---|
| Residue | Atom Name | Interaction Type | Distance (Å) |
| NH1 Arg 273 | O− (Pro2) | Salt bridge | 3.0 |
| OE1 Glu 375 | NH3+ (Ile) | Salt bridge | 4.1 |
| OE2 Glu 375 | NH3+ (Ile) | Hydrogen bond | 2.0 |
| NE2 His 378 | NH3+ (Ile) | Hydrogen bond | 2.4 |
| OE1 Glu 402 | NH3+ (Ile) | Hydrogen bond | 2.9 |
| OE2 Glu 402 | NH3+ (Ile) | Salt bridge | 3.1 |
| Ligand IIAE | |||
| NH2 Arg 273 | O1 (Glu) | Salt bridge | 3.0 |
| NH2 Arg 273 | OE1 (Glu) | Hydrogen bond | 2.9 |
| NH1 Arg 273 | OE2 (Glu) | Salt bridge | 3.0 |
| OE1 Glu 375 | NH3+ (Ile) | Salt bridge | 3.0 |
| OE2 Glu 375 | NH3+ (Ile) | Hydrogen bond | 2.6 |
| OE2 Glu 375 | NH (Ile) | Hydrogen bond | 2.4 |
| OE1 Glu 402 | NH3+ (Ile) | Hydrogen bond | 2.9 |
| OE2 Glu 402 | NH (Ala) | Hydrogen bond | 2.9 |
| CG Glu 402 | NH3+ (Ile) | Salt bridge | 4.3 |
| Ligand LIVTQ | |||
| ND1 His 374 | O (Gln) | Hydrogen bond | 2.8 |
| OE1 Glu 375 | NH3+ (Leu) | Salt bridge | 3.5 |
| OE2 Glu 375 | NH3+ (Leu) | Hydrogen bond | 2.9 |
| OE2 Glu 375 | NH (Ile) | Hydrogen bond | 2.3 |
| OE1 Glu 402 | NH3+ (Leu) | Hydrogen bond | 2.0 |
| OE2 Glu 402 | NH3+ (Leu) | Salt bridge | 4.3 |
| OE2 Glu 402 | NH (Val) | Hydrogen bond | 2.6 |
| OE1 Glu 406 | OH (Thr) | Hydrogen bond | 2.9 |
| OE1 Glu 406 | NH2 (Gln) | Hydrogen bond | 2.9 |
| OE1 Glu 406 | NH (Gln) | Hydrogen bond | 2.9 |
| OE2 Glu 406 | NH (Thr) | Hydrogen bond | 2.9 |
| OE2 Glu 406 | NH (Val) | Hydrogen bond | 2.3 |
| NE2 Gln 442 | O (Gln) | Hydrogen bond | 2.8 |
| NH2 Arg 518 | OH (Thr) | Hydrogen bond | 2.1 |
| Ligand LVYPFP | |||
| CG2 Thr 276 | O− (Pro) | Hydrogen bond | 2.6 |
| OE1 Glu 402 | NH3+ (Leu) | Salt bridge | 3.8 |
| OE2 Glu 402 | NH3+ (Leu) | Hydrogen bond | 2.9 |
| CO Glu 406 | NH3+ (Leu) | Salt bridge | 4.0 |
| OE1 Glu 406 | NH (Val) | Hydrogen bond | 2.8 |
| OE1 Glu 406 | NH (Tyr) | Hydrogen bond | 2.4 |
| OE2 Glu 406 | NH3+ (Leu) | Hydrogen bond | 2.8 |
In another molecular docking study conducted by Upreti et al. [58][73], chloroquine phosphate, a commercial ACE2 inhibitor, exhibited well-established hydrogen bonds with amino acid residues Glu 406, Asp 367, Asp 269, and Phe 274. Peptides LIVTQ and LVYPFP also interacted with the amino acid residue Glu 406 through hydrogen bonds and salt bridge interactions (Table 1; Figure 5 and Figure 6). Peptide LIVTQ additionally interacted with key residues His 374 and Glu 375 at distances of 2.8 Å and 3.5 Å, respectively (Table 1 and Figure 5). Moreover, Arg 273 is a key amino acid residue for substrate-binding in ACE2 that was found to form a salt-bridge with the C-terminal of the potent and selective human ACE2 inhibitor MLN-4760 [54][56][57][69,71,72]. Both peptides IPP and IIAE formed salt bridge interactions and hydrogen bonds with amino acid residue Arg 273 (Table 1; Figure 3 and Figure 4).
5. ACE and ACE2
Sequence alignment of ACE2 with ACE revealed the high conservation between these two enzymes. An analysis of the critical active site residues between ACE and ACE2 demonstrated that these two proteins are structurally well conserved. Since their active site structures are highly conserved and there exists a strong similarity between the catalytic domains of ACE and ACE2, consequently, the catalytic mechanism of ACE2 closely resembles that of ACE. However, due to differences in substrate specificity, distinctive key differences are present between the active site pockets of ACE and ACE2. Indeed, these differences occur in the ligand-binding pockets, particularly in the binding of the peptide C-terminal and at the S2′ subsite. The cavity in ACE2 is smaller than that of ACE, which allows an extra amino acid to bind in the specificity pocket. Additionally, the S1 subsite of ACE is larger than that of ACE2 [55][70]. In our previous work, we provided strong in vitro evidence for the ACE inhibitory activity of peptide sequences IPP, IIAE, LIVTQ, and LVYPFP [52][53][67,68]. Although all four peptides have been found to exhibit high ACE inhibitory activity, peptide IIAE formed strong hydrogen bonds with the amino acid residues Gln 259 and Thr 358 in the active site of ACE, in common the ACE inhibitory drug Sampatrilat. IIAE also interacted with the amino acid residues Gln 259 and His 331, in common with other ACE-inhibitory drugs such as Captopril, Lisinopril, and Elanapril, and with the amino acid residue Asp 140, in common with Lisinopril. Additionally, IPP has been identified as the most potent ACE inhibitor from milk protein [59][74]. When compared to the other peptides’ interactions with ACE, IIAE and IPP seem to display the highest ACE inhibitory activities. Notably, according to the ACE2 docking results, both peptides IPP and IIAE also seem to display the highest ACE2 inhibitory activity.
6. Potential Use of ACE Inhibitors in the Treatment of COVID-19
To date, there is no effective drug available to treat COVID-19 patients [60][75]. Although COVID-19 vaccines were shown to be closely associated with a significant reduction in symptomatic infections [61][76], vaccine hesitancy is widespread worldwide, which could hinder populations from achieving herd immunity [62][77]. The rapid global emergence of novel SARS-CoV-2 variants, the unequal international distribution of COVID-19 vaccines, and slow vaccine rollouts, especially in developed countries, could also be significant factors obstructing the achievement of herd immunity and the end of the pandemic. Although antimalarial drugs chloroquine and hydroxychloroquine, and some synthetic drugs such as remdesivir [60][75] and ritonavir/lopinavir [60][63][75,78] are currently used to treat COVID-19 patients, currently, there remains no effective and approved drug available against COVID-19 [64][79]. Various side effects associated with the aforementioned drugs were also observed among treated patients [60][63][75,78], delaying widespread acceptance and administration.
Consequently, identifying safe and effective compounds that can restrain the entry of SARS-CoV-2 into host cells via ACE2 is a priority for the scientific community. In this respect, an active area of research is the impact of milk/whey-derived bioactive peptides and their potential health benefits as ingredients of health-promoting functional foods [65][80]. Peptide sequences from whey proteins exhibit different bioactivities, including ACE inhibitory activity. In fact, milk is the main source of antihypertensive ACE-inhibitory peptides reported to date [48][66][63,81].
In the scientific community, controversy has arisen regarding whether the use of ACE inhibitors would be harmful or beneficial in the context of the COVID-19 pandemic [33][34][35][67][68][69][48,49,50,82,83,84]. Although increased COVID-19 disease severity seems to manifest in people with cardiovascular comorbidities [21][70][37,85], it is suggested that this association could be related to advanced age and obesity [70][85]. Moreover, there seems to be growing evidence that the use of ACE inhibitors does not worsen the prognosis of COVID-19 [71][86]. In fact, in a cohort study including 8.3 million people, ACE inhibitors were not found to be significantly associated with increased risks of COVID-19 disease, nor of requiring ICU care [42][57]. In agreement, another meta-analysis study also reported that the use of ACE inhibitors was not associated with requiring intensive care, mechanical ventilation, progression to severe disease, and increased risk of death. However, some researchers have reported a 16% reduction in the risk of COVID-related mortality with the use of ACE inhibitors [68][83].
Some studies suggest that ACE inhibitors could even play a protective role in hypertensive patients by averting organ injury [72][87]. Indeed, in vivo models support the role of ACE inhibitors in blunting lung injury and exerting health benefits in both human and animal trials [73][74][75][76][88,89,90,91]. Data from human studies also revealed that ACE inhibitors can reduce or prevent pneumonia [77][78][92,93]; specifically, the (i) treatment of chronic obstructive pulmonary disease with ACE inhibitors was found to reduce disease complications and (ii) treatment with ACE inhibitors was shown to mitigate the effects of radiation pneumonitis [79][94]. In short, there is consistent evidence indicating that ACE inhibitors seem to have beneficial effects in modulating lung damage, including in the context of pulmonary injury caused by viral infection. Due to insufficient evidence of the potentially harmful effects of ACE inhibitors and considering the overwhelming evidence supporting their benefits, multiple scientific societies rejected the recommendation to discontinue ACE inhibitors in the context of the COVID-19 pandemic [80][81][82][95,96,97]. Interestingly, ACE inhibitors were reported to be associated with significant pulmonary inflammatory response reductions in patients admitted with viral pneumonia [83][98] and attenuated inflammatory response in COVID-19-infected patients [84][85][99,100]. This emerging evidence prompted many researchers to advocate for the use of RAS inhibitors in the therapeutic management of COVID-19 infection [68][83].
Regarding the role of the RAS pathway in the pathophysiology of COVID-19 and SARS-CoV-2 infection, there are two primary theories. First, data from the literature have shown that Ang II-mediated inflammation is a main mediator of acute lung injury and fibrosis [30][86][87][45,101,102]. Similar to SARS-CoV, loss of ACE2 activity and expression could lead to an increase in Ang II levels in the lungs and could consequently induce COVID-19 acute lung injury. One study reported significantly higher Ang II levels in COVID-19 patients that correlated with viral load and indicators of lung injury. However, this study had considerable methodological limits: only 12 patients took part in the clinical study and circulating levels of ACE and/or ACE2 were not determined [1][38][88][5,53,103]. Furthermore, data from the original SARS-CoV epidemic indicated that infection with SARS-CoV-2 may lead to ACE-2 dependent myocardium infection, which results in decreased cardiac ACE2 expression, accelerating acute heart injury [89][104]. However, it is important to note that there is no clinical data to confirm this.
Second, there is concern that ACE inhibitors may potentially increase the expression and levels of ACE2 in the lungs, which facilitates SARS-CoV-2 infection such that administering ACE inhibitors may increase the risk of severe and fatal disease [37][90][52,105]. In select animal models, ACE inhibitors were able to increase heart and kidney ACE2 expression [36][91][51,106]. However, there are no data proving that these compounds can increase lung ACE2 expression in both animal models and human trials. In a similar manner, there are no available data demonstrating that the increased expression of ACE2 would necessarily indicate an increased risk of disease severity or infection, or that the use of these agents is correlated with increased virulence or viral infectivity. In fact, there does not appear to be any consistent association between increased ACE2/Ang (1–7)/Mas pathway activity and expression, and the use of ACE inhibitors in the few clinical studies assessing the effect of ACE inhibitors on the ACE2/Ang (1–7) pathway [92][93][94][95][107,108,109,110]. Although there is a lack of evidence to demonstrate the effect of ACE inhibitors on ACE2 expression and thus SARS-CoV-2 infectivity, the bulk of the experimental evidence indicates that ACE inhibitors may reduce the action of Ang II and consequently attenuate Ang II-driven acute lung injury [38][39][53,54]. ACE inhibitors therefore offer promise as potential novel therapeutics to treat COVID-19 disease [31][46].
Intriguingly, based solely on experimental studies in which RAS inhibitors were administered in vivo [96][97][98][99][100][111,112,113,114,115], Zamai, 2020 [101][116] highlighted a reasonable hypothesis, in which he stated that using inhibitors which block both ACE2 and ACE pathways in COVID-19 patients could be very beneficial in the treatment of COVID-19. In short, observations from these studies indicate that hypoxia/hypercapnia, a condition that occurs in SARS patients, is highly likely to upregulate the activity of both arms of the RAS. A strong correlation was also observed between the gene expression of ACE2 and that of ACE [102][117]. Another observation suggested the possibility of a positive feedback induced by SARS-CoV infection, leading to the surface expression of both ACE and ACE2 [103][104][105][118,119,120]. Altogether, these observations indicate that RAS-mediated positive feedback loops can be induced by SARS-CoV-2 at different organ levels. Consequently, in order to block these feedback loops, Zamai, 2020 [101][116] suggested that different compounds can be produced to inhibit RAS pathways and subsequently to prevent critical, advanced, and untreatable stages of the COVID-19 disease.
IPP, IIAE, LIVTQ, and LVYPFP, which are bioactive peptides derived from whey proteins, were initially characterized as ACE inhibitors through in vitro and in silico assays in our previous works [52][53][67,68]. Findings from the current study demonstrate additional novel effects for these bioactive whey-derived peptides as potential ACE2 inhibitors. These results strongly support our hypothesis that these whey-derived peptides not only could exhibit ACE inhibitory activity but also could bind to ACE2 and, as such, could have a potential effect of intervening in the interaction between the ACE2 and SARS-CoV-2 S protein. Additionally, compared to synthetic ACE-inhibitory drugs, these peptides are from a natural source and do not exhibit toxic side effects, which might also help to reduce the risks associated with traditional drugs in the treatment of COVID-19 infection.