Exosomes are attractive as vehicle systems for small therapeutic molecules and/or biomolecules including nucleic acids and proteins because of their lipid nature, presence of specific surface ligands (CD11b and CD18 receptors, integrins, tetraspanins) and ability to cross the blood–brain barrier. When compared to other drug delivery systems, exosomes have the distinct advantages of blood–brain barrier penetrance, longer duration in systemic circulation, tissue specificity that minimizes unwanted toxicity or off-target effects, stability of content, desirable biocompatibility and minimal toxicity issues. Techniques such as fusion expression, exosome membrane surface display and anchoring platforms have been used to attach peptides and biological ligands of interest to adhesion molecules, tetraspanins or integrins on exosome surface to ensure targeted delivery and enhanced uptake into desired cells.
- extracellular vesicle
- exosome
- CNS
- neuromuscular disease
- neurodegenerative disease
- biomarkers
1. Introduction
Mechanisms involving chemical messengers, the extracellular matrix, gap junctions, tunnelling nanotubes and extracellular vesicles exist in cells for communication and exchange of bioactive materials including organelles, genetic materials, pathogens and misfolded proteins [1]. Based on characteristics of their production and release from cells, classes of extracellular vesicles include exosomes, microvesicles and apoptotic bodies [2]. The use of the term “exosomes” can be traced to a 1981 paper where the writers proposed that exfoliated membrane vesicles be referred to as exosomes [3]. In 1983, two independent studies reported that the maturation of reticulocytes into erythrocytes involved the release of transferrin receptors via 50 nm vesicles [4][5][4,5]. Four years later, the term exosome was used by Rose Johnstone to refer to vesicles released into the extracellular space following fusion of the multivesicular bodies (MVBs) with the plasma membrane [6]. For the purpose of this review, and in line with the International Society for Extracellular Vesicles (ISEV) designation [7], the term small EVs (sEVs) will be used interchangeably with exosomes. Exosomes can be released by different cell types in vitro and can be detected in biological fluids in both pathological and physiological contexts.
Intercellular communication between neurons [8] and their surrounding cells [9][10][11][9,10,11] in the central nervous system occurs through the secretion of soluble molecules or release of vesicles such as exosomes containing neuroprotective factors in the extracellular space [12][13][12,13], participating in the maintenance of the brain homeostasis [14][15][14,15]. Under neurodegenerative conditions associated with ageing, such as amyotrophic lateral sclerosis (ALS), Alzheimer’s or Parkinson’s disease, exosomes are suspected to propagate toxic proteins [16][17][18][19][16,17,18,19].
After summarizing exosome biogenesis, exosome heterogeneity and their fate and impact on recipient cells, this review will then focus on the role of sEVs in Amyotrophic Lateral sclerosis (ALS), a multisystemic condition [20]. Thereafter, we will discuss whether exosomes could be used as biomarkers for ALS.
2. Exosome Biogenesis and Secretion
During exosome biogenesis, early endosomes mature into late endosomes where intralumenal vesicles (ILVs) are formed and accumulate in their lumen. The process of exosome formation includes (1) the clustering of sorted cargo at the membrane of the MVBs, forming microdomains, and (2) subsequent membrane curvature and fission of vesicles. Generally, the fate of MVBs is to fuse with lysosomes for degradation of their content. However, MVBs can also be targeted to the plasma membrane of the cell where ILVs are released into the extracellular space as exosomes upon membrane fusion [21]. The role of endosomal sorting complexes required for transport (ESCRT) proteins in exosome biogenesis has been investigated using a variety of approaches such as proteomics and RNA silencing screening analysis [22][23][22,23]. The depletion of the four ESCRT complexes involved in exosome biogenesis did not totally abrogate exosome formation, indicating the existence of other mechanisms [23][24][23,24]. Two different pathways are described for sEV formation ( Figure 1 ), summarised below.
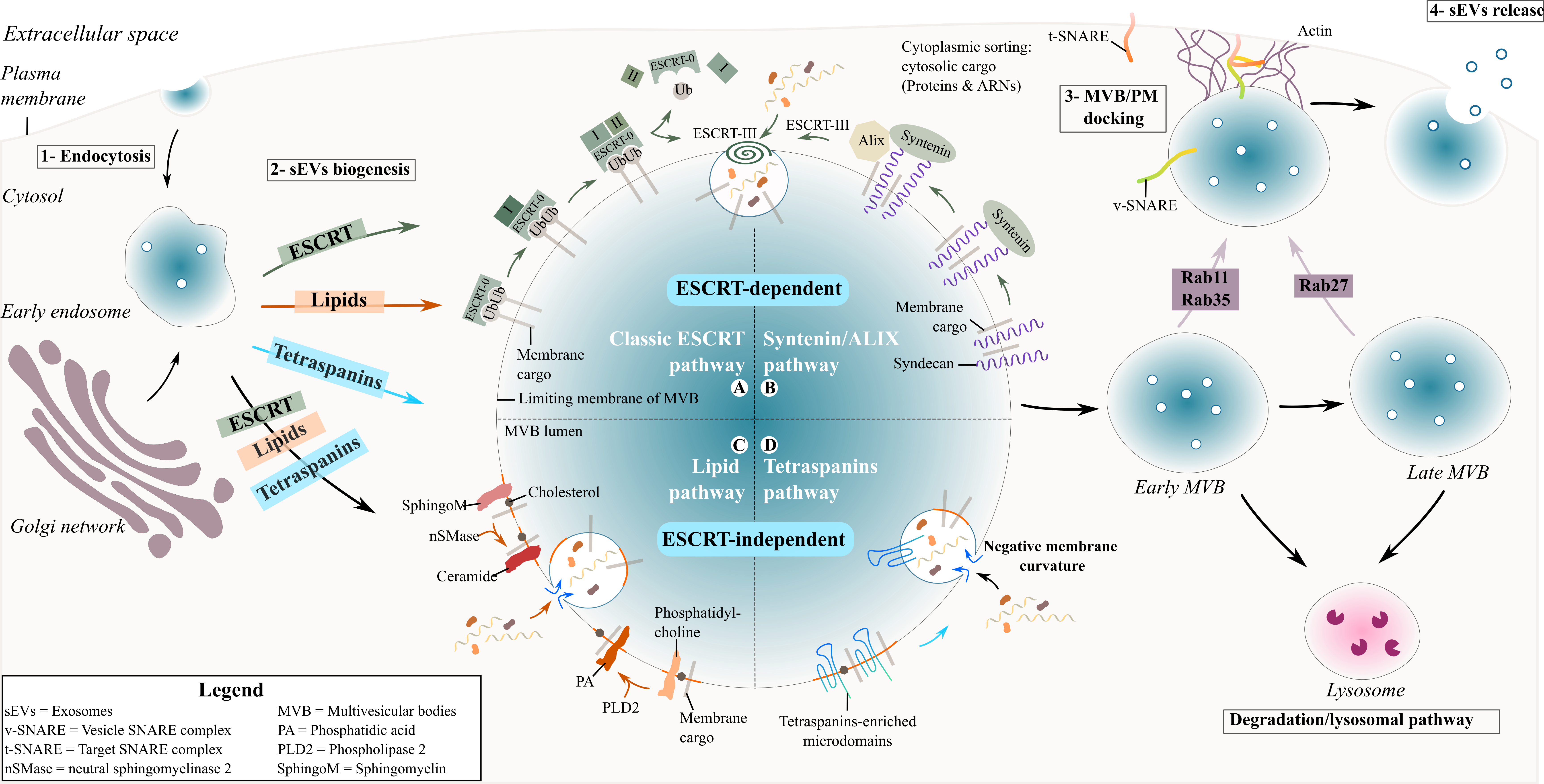 Figure 1. Biogenesis and secretion of exosomes. Schematic representation of exosome formation and release in the extracellular space.
Figure 1. Biogenesis and secretion of exosomes. Schematic representation of exosome formation and release in the extracellular space.
The ESCRT proteins can cluster into four complexes: ESCRT-0, I, II and III [25] and are involved in the sorting of ubiquitinated cargo into ILVs ( Figure 1 ). The ESCRT-0 complex is composed of HRS (Hepatocyte growth factor-regulated tyrosine kinase substrate) and STAM (Signal transducing adapter molecule) proteins and is recruited to the endosomal membrane via ubiquitinated cargo and phosphatidylinositol 3-phosphate (PI3P). HRS recognizes ubiquitinated protein–ubiquitin acting in this context as a targeting signal for the specific incorporation of molecules in ILVs- and binds to PI3P [26][27][26,27]. The HRS/STAM complex recruits ESCRT-I via TSG101/VPS28 (two components of the ESCRT-I complex) to the endosomal membrane and forms an ESCRT-0/ESCRT-I complex. The ESCRT-I complex contains one copy each of TSG101, VPS28, VS37 [26] and MVB12. Its recruitment at the endosomal membrane is enhanced by ubiquitinated transmembrane cargo. Similar to ESCRT-0, it is also involved in the clustering of selected ubiquitinated cargo into microdomains and mobilizes the ESCRT-II complex. The ESCRT-II complex is a heterodimer comprising one copy each of VPS36 and VPS22 and two VPS25 subunits [28]. Together with the ESCRT-I complex, the ESCRT-II complex initiates the negative curvature of the emerging ILV at the MVB membrane and the uptake of cytosolic cargo [27]. Finally, the association of ESCRT-I and –II recruits the ESCRT-III complex at the ILV biogenesis site via ALIX or through a direct interaction with VPS25 from ESCRT-II. The components of the ESCRT-III complex polymerize into filaments after recruitment at the MVB membrane with two protein complexes, VPS2-VPS24 and VPS20-SNF7 [28][29][28,29]. ESCRT-III inside the nascent neck of the ILV leads to the closure and detachment of vesicles containing specific cargo within the MVB lumen [26][27][26,27].
Another ESCRT-mediated exosome biogenesis involves the interaction of ESCRT-III/ALIX with the transmembrane proteoglycan receptor, syndecan, and its binding partner syntenin [30]. Syndecans assemble at the MVB membrane, followed by cleavage of the syndecan auto-repulsive domain. They remain clustered at the membrane allowing syntenin to bind to the syndecan bundle. Consequently, syntenin interacts with ALIX, recruiting the ECRT-III unit with VPS4 and leading to endosomal membrane inward budding and abscission. ALIX initiates a de-ubiquitination step that occurs before the incorporation of proteins into the ILV and before the closure of the latter [26].
The tetraspanin family are regulators of non-ESCRT dependent exosome biogenesis ( Figure 1 ). Tetraspanin proteins possess four transmembrane domains resulting in two extracellular and three intracellular regions [31][33]. Tetraspanin proteins are glycosylated to various degrees [31][33], forming oligomers and a protein-enriched microdomain at the plasma membrane [32][34]. The role of glycosylation modifications of tetraspanins is still unknown but it possibly contributes to tetraspanin complex formation [33][35]. Due to their cone-shaped conformation and their ability to cluster into microdomains, tetraspanins could induce inward budding of the late endosomal membrane and exosome formation [34][36]. Tetraspanins are highly enriched within exosomes and have been specifically used as exosomal biomarkers over the years (see Table 1 ). Among them CD63, CD81, CD82 or CD9 are particularly used as exosome markers.
Table 1. Exosomes released by cells in the central nervous system and by the neuromuscular system.
|
Exosome origin |
ALIX |
CD9 |
CD63 |
CD71 |
CD81 |
CD82 |
Flotillin-1 |
Hsp70 |
Hsp90 |
Lamp1 |
Lamp2 |
Rab7 |
Rab11 |
Tsg101 |
Calnexin |
Ref |
|
Cortical neurons |
+ |
+ |
|
+ |
|
|
+ |
|
[8,37-39] |
|||||||
|
Microglial cell |
+ |
+ |
+ |
+ |
|
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
[11,40-50] |
||
|
Oligodendrocytes |
+ |
+ |
|
+ |
+ |
- |
||||||||||
|
Schwann cells |
+ |
+ |
+ |
|
|
+ |
+ |
+ |
|
+ |
[54–59] |
|||||
|
Astrocytes |
+ |
|
+ |
|
+ |
+ |
+ |
+ |
[9,60–66] |
|||||||
|
Hippocampal neurons |
+ |
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
[67] |
|
Motor neurons |
+ |
|
+ |
|
|
|
+ |
|
|
|
|
|
|
|
|
[68] |
|
Skeletal muscle cells |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
|
|
|
|
+ |
- |
[69–79] |
3. Exosomal Interaction with Recipient Cells and the Fate of Exosomes
Understanding whether exosomes communicate with the recipient cell in a specific and controlled or stochastic process is the first step in unravelling exosome-cell interaction [35][36][125,126]. Intercellular communication mediated by secreted exosomes occurs either by direct interaction with recipient cell with or without the uptake of exosomes and/or indirect interaction facilitated by cleaved transmembrane ligands (proteins or lipids). This section addresses the types of interactions between exosomes and the recipient cell ( Figure 2 ).
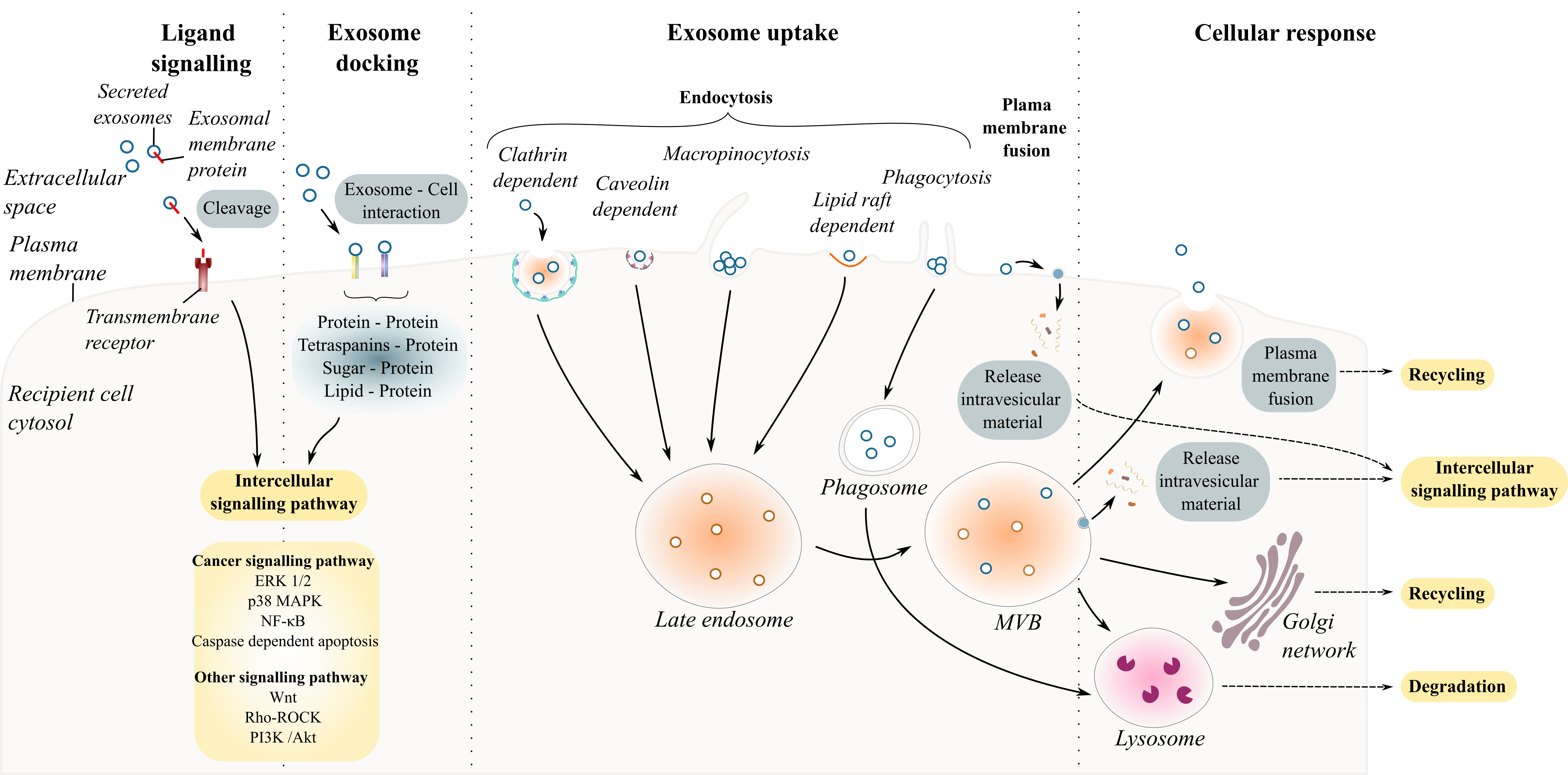 Figure 2. Exosome and recipient cell communication. Schematic diagram summarizing exosome–cell interactions. Exosome interaction with recipient cells can occur through the endocytic pathways including clathrin- or caveolin- dependent endocytosis, macropinocytosis, phagocytosis, and lipid-raft mediated endocytosis.
Figure 2. Exosome and recipient cell communication. Schematic diagram summarizing exosome–cell interactions. Exosome interaction with recipient cells can occur through the endocytic pathways including clathrin- or caveolin- dependent endocytosis, macropinocytosis, phagocytosis, and lipid-raft mediated endocytosis.
Exosome–recipient cell interaction requires a combination of specific molecules present on the surfaces of the cell and on the exosome including proteins (glycoproteins, integrins or tetraspanins), sugar (heparan sulfate proteoglycans) and lipids. Table 2 reports some of the ligands on the surface of exosomes and the targeted cells that are known to interact with each other.
Table 2. Summary of ligand–receptor interaction in exosomes/cell communication.
| Exosome ligand | Target cell ligand | Ref | |
| Glycoproteins | Fibronectin | Heparin sulfate proteoglycans (HSPGs) | [129] |
| Fibronectin | Integrins | [130] | |
| ICAM (CD54) | LFA-1 | [131–133] | |
| MUC1 | DC-SIGN | [134] | |
| Integrins | β1 and β2 integrins | ICAM-1 | [135] |
| β1 and β2 integrins | Collagen-I | ||
| β1 and β2 integrins | Fibronectin | ||
| Integrin α4β1 | Fibronectin | [136] | |
| αvβ3 / αvβ5 integrins | MFG-E8 | [137] | |
| CD47 | SIRPα | [138] | |
| Lectin | C-type lectin | Mannose-rich C-type lectin receptor | [139] |
| Galectin 5 | Glycoproteins (CD7, α5β1-integrin, or laminin) | [140] | |
| Galectin 9 | Tim 3 | [141] | |
| Tetraspanins | Tspan8-CD49d | ICAM-1 (CD54) | [142] |
| Lipid rafts | Phosphatidylserine | Tim-1/4 | [54] |
| Phosphatidylserine | MFG-E8 | [137] | |
| Phosphatidylethanolamine | MFG-E8 | ||
| Annexin 2 | Lipid raft domain | [143] | |
| Sugar | α2,3-linked sialic acids | sialoadhesin (CD169) | [144] |
Several studies have investigated how exosomes could specifically target different cells under physiological and pathological conditions [37][38][39][136,143,144]. Among the list of known ligand– receptor interactions, protein–protein interactions are the most abundant for sEVs. For example, the pre-treatment of ovarian cell-derived exosomes with proteinase K or trypsin to degrade exosomal transmembrane protein abolished their uptake by cancer cells [37][40][41][136,145,146]. Inhibiting specific interactions using antibodies or soluble ligands prior to treatment of cells with exosomes has enabled the discovery of many specific ligand–receptors involved in exosome uptake [42][147]. The various specific ligand–receptors suggest that some sEVs may target specific cells and/or may have different effects on different cells. Once an exosome docks at the surface of the recipient cells, four scenarios can occur. The exosome binds to cell surface receptors eliciting intracellular responses in the recipient cell [43][148]; it fuses with the plasma membrane releasing its contents directly into the cytosol [44][149]; it is internalized by the recipient cell via the endosome machinery [45][46][150,151]; or it crosses the cell and is re-released intact to target other cell types [47][152].
4. Exosomes as Molecular Biomarkers for ALS
Presently, no single diagnostic test exists for the diagnosis of ALS, with clinicians relying on a combination of history, physical examination, neuroimaging, electrodiagnostic and laboratory findings [20][48][20,222]. ALS shares certain overlap with other neurodegenerative diseases, which makes diagnosis difficult with a reported lag of 12 months between the onset of symptoms and neurological diagnosis [48][222]. Biomarkers provide opportunities to improve diagnosis, monitor disease progression, gauge prognosis, aid patient stratification and response to therapy [20][49][20,273], and exosomes may be well suited for these roles. Molecular biomarker development for ALS is at an all-time high with investigated biomarkers cutting across proteins, miRNAs, mRNAs and metabolites from cerebrospinal fluid (CSF) and blood (extensively reviewed in [20]).
The investigation of sEVs as diagnostic and/or prognostic biomarkers for ALS has been increasingly investigated during the last decade. Disease pathology affects the composition of exosomes [50][274] as well as their secretion and/or accumulation [51][52][275,276]. Advantageously, proteins and RNAs associated with a disease and enclosed in exosomes exhibit stability in biological fluids as they are protected from degradation by the double membrane structure of the sEVs and can be stored for long periods before analysis [53][54][277,278]. This would suggest that the content could be sensitively detected if appropriate isolation protocols that ensure near-pure sEVs are utilised. Exosomes and their contents as molecular biomarkers for ALS have been investigated in cerebrospinal fluid [279,280], plasma ([281–283] and serum [284].
References
- Nawaz, M.; Fatima, F. Extracellular vesicles, tunneling nanotubes, and cellular interplay: Synergies and missing links. Front. Mol. Biosci. 2017, 4, 50. Julien Fauré; G. Lachenal; M. Court; J. Hirrlinger; C. Chatellard-Causse; B. Blot; J. Grange; G. Schoehn; Yves Goldberg; V. Boyer; et al.Frank KirchhoffG. RaposoJ. GarinR. Sadoul Exosomes are released by cultured cortical neurones. Molecular and Cellular Neuroscience 2006, 31, 642-648, 10.1016/j.mcn.2005.12.003.
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. Mathilde Chivet; Charlotte Javalet; Karine Laulagnier; Béatrice Blot; Fiona J. Hemming; Rémy Sadoul; Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. Journal of Extracellular Vesicles 2014, 3, 24722, 10.3402/jev.v3.24722.
- Trams, E.G.; Lauter, C.J.; Salem, N.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 645, 63–70. Karine Laulagnier; Charlotte Javalet; Fiona J. Hemming; Rémy Sadoul; Purification and Analysis of Exosomes Released by Mature Cortical Neurons Following Synaptic Activation. Methods in Molecular Biology 2016, 1545, 129-138, 10.1007/978-1-4939-6728-5_9.
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. Yaying Song; Zongwei Li; Tingting He; Meijie Qu; Lu Jiang; Wanlu Li; Xiaojing Shi; Jiaji Pan; Linyuan Zhang; Yongting Wang; et al.Zhijun ZhangYaohui TangGuo-Yuan Yang M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910-2923, 10.7150/thno.30879.
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978.
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420.
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648.
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4.
- Krämer-Albers, E.-M.; Bretz, N.; Tenzer, S.; Winterstein, C.; Möbius, W.; Berger, H.; Nave, K.-A.; Schild, H.; Trotter, J. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteomics. Clin. Appl. 2007, 1, 1446–1461.
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic Analysis of Microglia-Derived Exosomes: Metabolic Role of the Aminopeptidase CD13 in Neuropeptide Catabolism. J. Immunol. 2005, 175, 2237–2243.
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte-Neuron Communication. PLoS Biol. 2013, 11, e1001604.
- Fröhlich, D.; Kuo, W.P.; Frühbeis, C.; Sun, J.J.; Zehendner, C.M.; Luhmann, H.J.; Pinto, S.; Toedling, J.; Trotter, J.; Krämer-Albers, E.M. Multifaceted effects of oligodendroglial exosomes on neurons: Impact on neuronal firing rate, signal transduction and gene regulation. R. Soc. 2014, 369, 20130510.
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53 (Suppl. 6), 1–6.
- Zagrean, A.-M.; Hermann, D.M.; Opris, I.; Zagrean, L.; Popa-Wagner, A. Multicellular Crosstalk Between Exosomes and the Neurovascular Unit After Cerebral Ischemia. Therapeutic Implications. Front. Neurosci. 2018, 12, 811.
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced -Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851.
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759.
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117.
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.-R. A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 2019, 10, 400.
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312.
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318.
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565.
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular Endosome Biogenesis in the Absence of ESCRTs. Traffic 2009, 10, 925–937.
- Babst, M. A Protein’s Final ESCRT. Traffic 2005, 6, 2–9.
- Williams, R.L.; Urbé, S. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 2007, 8, 355–368.
- Kalra, H.; Drummen, G.; Mathivanan, S.; Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170.
- Hanson, P.I.; Cashikar, A. Multivesicular Body Morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362.
- Babst, M.; Katzmann, D.J.; Estepa-Sabal, E.J.; Meerloo, T.; Emr, S.D. ESCRT-III: An endosome-associated heterooligomeric protein complex required for MVB sorting. Dev. Cell 2002, 3, 371–382.
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685.
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648.
- Charrin, S.; Manié, S.; Thiele, C.; Billard, M.; Gerlier, D.; Boucheix, C.; Rubinstein, E. A physical and functional link between cholesterol and tetraspanins. Eur. J. Immunol. 2003, 33, 2479–2489.
- Termini, C.M.; Gillette, J.M. Tetraspanins Function as Regulators of Cellular Signaling. Front. Cell Dev. Biol. 2017, 5, 34.
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.M.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051.
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045.
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345.
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503.
- Hung, W.-T.; Navakanitworakul, R.; Khan, T.; Zhang, P.; Davis, J.S.; McGinnis, L.K.; Christenson, L.K. Stage-specific follicular extracellular vesicle uptake and regulation of bovine granulosa cell proliferation. Biol. Reprod. 2017, 97, 644–655.
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69.
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108.
- Bretz, N.P.; Ridinger, J.; Rupp, A.-K.; Rimbach, K.; Keller, S.; Rupp, C.; Marmé, F.; Umansky, L.; Umansky, V.; Eigenbrod, T.; et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J. Biol. Chem. 2013, 288, 36691–36702.
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641.
- Hakulinen, J.; Junnikkala, S.; Sorsa, T.; Meri, S. Complement inhibitor membrane cofactor protein (MCP; CD46) is constitutively shed from cancer cell membranes in vesicles and converted by a metalloproteinase to a functionally active soluble form. Eur. J. Immunol. 2004, 34, 2620–2629.
- Prada, I.; Meldolesi, J. Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. Int. J. Mol. Sci. 2016, 17, 1296.
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267.
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes Derived from Epstein-Barr Virus-Infected Cells Are Internalized via Caveola-Dependent Endocytosis and Promote Phenotypic Modulation in Target Cells. J. Virol. 2013, 87, 10334–10347.
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Götz, J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 2018, 6, 10.
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58.
- Morgan, S.; Duguez, S.; Duddy, W. Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. J. Pers. Med. 2018, 8, 44.
- Kim, Y.S.; Ahn, J.S.; Kim, S.; Kim, H.J.; Kim, S.H.; Kang, J.S. The potential theragnostic (diagnostic+therapeutic) application of exosomes in diverse biomedical fields. Korean J. Physiol. Pharmacol. 2018, 22, 113–125.
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650.
- Candelario, K.M.; Steindler, D.A. The role of extracellular vesicles in the progression of neurodegenerative disease and cancer. Trends Mol. Med. 2014, 20, 368–374.
- Boukouris, S.; Mathivanan, S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom.-Clin. Appl. 2015, 9, 358–367.
- Jin, Y.; Chen, K.; Wang, Z.; Wang, Y.; Liu, J.; Lin, L.; Shao, Y.; Gao, L.; Yin, H.; Cui, C.; et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 2016, 16, 753.