Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Bruce Ren and Version 1 by Saravannan Sekaran.
Tissue-nonspecific alkaline phosphatase (TNAP) is a key enzyme responsible for skeletal tissue mineralization. It is involved in the dephosphorylation of various physiological substrates, and has vital physiological functions, including extra-skeletal functions, such as neuronal development, detoxification of lipopolysaccharide (LPS), an anti-inflammatory role, bile pH regulation, and the maintenance of the blood brain barrier (BBB). TNAP is also implicated in ectopic pathological calcification of soft tissues, especially the vasculature. Although it is the crucial enzyme in mineralization of skeletal and dental tissues, it is a logical clinical target to attenuate vascular calcification.
- alkaline phosphatase
- bone
- mineralization
- brain
- vasculature
- calcification
1. Introduction
Skeletal tissues are extraordinary structures and their biomechanical strength is attributed to the orchestrated process of biomineralization-an intricately controlled event involving the cell driven deposition of hydroxyapatite from ions largely present in body fluids [1]. Physiological mineralization is observed in hard tissues [2,3][2][3] and pathological mineralization is widely observed in soft tissues [4,5,6,7,8,9][4][5][6][7][8][9]. Mineralized extracellular matrix is a unique feature of the vertebral system in animals. Bone is a multifaceted organ undergoing remodeling throughout the lifetime by balanced actions of osteoblasts, osteoclasts and osteocytes. Hydroxyapatite (HAP) mineral is hierarchically organized on the type I collagen matrix [10,11][10][11]. Osteoblasts are responsible for the laying of organic mineralized matrix. They secrete type 1 collagen which is templated for mineral nucleation and subsequent crystal growth. HAP nucleation results in growth of the crystal following a continuous cross-fibrillar pattern [12,13,14,15][12][13][14][15]. The hierarchical arrangement of bone structure and model of collagen microfibril along with HAP arrangement is depicted in Figure 1. The process of mineralization is involving direct mechanism mediated by extracellular vesicles (EV’s) released from osteoblasts and an indirect mechanism in which non collagenous proteins that are negatively charged, associate with collagen and direct various mineral precursors for nucleation [16,17][16][17]. As a result of the mechanical properties of mineralized tissues, they act as a depot for various minerals that are essential for numerous physiological processes in the body. Alkaline phosphatases (E.C. 3.1.3.1) regulate mineralization in hard tissue under both physiological and pathological conditions. Moreover, they catalyze the dephosphorylation of various physiological and non-physiological substrates [18].
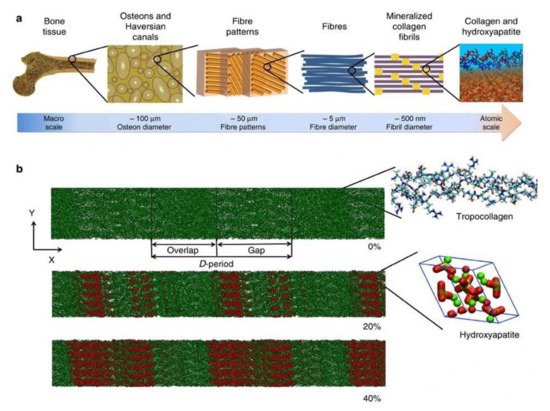
Figure 1. (a) Hierarchical structure of bone depicting the arrangement from macro to nanoscale. (b) Collagen microfibril model indicating different levels of mineralization from 0% to 40% indicating the alignment of hydroxyapatite crystals. The c-axis of HAP aligns with the collagen fibril axis. Green color indicates Ca atoms, red and white denote OH groups and phosphate groups are visualized in the tetrahedron structure. The image was taken from Arun et al. [19].
2. TNAP
Tissue-nonspecific alkaline phosphatase (TNAP) is highly expressed on the plasma membrane of osteoblasts, odontoblasts and hypertrophic chondrocytes and also highly concentrated in the extracellular vesicles (EVs) originating from these cells [33,34][20][21]. Pyrophosphate (PPi) and pyridoxal phosphate (PLP), phosphoethanolamine (PEA) are believed to be the physiological substrates [35,36][22][23]. TNAP catalyzes the hydrolysis of PPi, an inhibitor of bone mineralization and provides inorganic phosphate ions for hydroxyapatite nucleation and formation [37,38,39][24][25][26]. PLP is essential for the formation of neurotransmitters such as dopamine, histamine, serotonin, taurine and Gamma-aminobutyric acid (GABA) [40][27]. Pyridoxal administration to Alpl−/− mice prevents epileptic seizures indicating its role in PLP metabolism. PLP entry into the cells is mediated by TNAP which dephosphorylates PLP into pyridoxal and again transformed back into PLP in neurons [41,42][28][29]. Unlike PPi and PLP, the confirmed role of phosphoethanolamine as a substrate for TNAP is not known but TNAP deficient patients and knockout mice display elevated levels of phosphoethanolamine [43][30]. Recent reports also depict that di-phosphoryl lipopolysaccharide (LPS) [44[31][32],45], adenosine triphosphate (ATP) [46,47,48][33][34][35] and phosphorylated osteopontin (pOPN) [49,50,51][36][37][38] are TNAP natural substrates.
2.1. Gene Structure
Four human ALP isoenzymes are present. TNAP is ubiquitously expressed in bone, liver, kidney, white blood cells and neuronal cells. Human TNAP is encoded by ALPL gene (NCBI Gene ID: 249) which is located on the chromosome 1 short arm (1p36.1-34). The ALPL gene possesses 1.5 kb of coding region, spans more than 50 kb of genomic DNA and contains 12 exons, including 11-proetein coding exons with 2536 transcript length [34,52][21][39]. The promoter region of ALPL gene contains a TATA box, Sp1 binding site and retinoic acid responsive element (RARE) which regulates TNAP gene expression [53,54][40][41]. The regulation of TNAP expression by retinoic acid is through RARE and active vitamin D is regulated by modifying the stability of TNAP mRNA [55][42]. In addition to this, phosphates produced by the enzymatic activity of ALP are also known to regulate TNAP expression [56][43]. The methylation of promoter regions of the ALPL gene is known to be involved in epigenetic regulation [57][44]. Genes encoding tissue-specific ALPs are present on chromosome 2 long arm and possess a compact gene arrangement [26,58,59][45][46][47].
2.2. Protein Structure
TNAP is an ectoenzyme approximately 80kDa, linked to the outer plasma membrane via glycosylphosphatidylinositol (GPI) anchorage [60][48]. The GPI consists of an ethanolamine phosphate and three glucosamine, mannose, and phosphatidylinositol. It can be sliced by phospholipases present in plasma membranes explaining the circulatory presence of TNAP in various biological fluids [61][49]. TNAP is synthesized initially as a 66 kDa peptide followed by the addition of O-and N-glycosides in the ER. Among mammalian ALPs 58% of aminoacid residues are highly conserved in human TNAP sequences [62][50]. Until now, the 3D structure of TNAP is not yet delineated and a simulation-based model of mouse IAP or human PLAP is used to describe the structure (Figure 2). The presence of a catalytic serine residue (S92) is marked in the active site of human PLAP and it also contains two Zn2+ binding sites and one Mg2+ binding site. A Ca2+ binding site is also seen in human and murine TNAP but it does not possess any direct role on the catalytic activity. The crown region is present in mammalian ALPs and known to interact with various extracellular proteins including type 1 collagen [63][51]. Isoforms of TNAP are distinguished by their distribution pattern on electrophoresis owing to the difference in O-linked sugar chains [60][48]. Dimerization of two TNAP monomers is mediated by two disulfide bonds that play a prominent role in the severity of hypophosphatasia. TNAP contains five possible N-linked glycosylation sites (N123, 213, 254, 286 and 413) with sugar chains essential for its catalytic activity and types of sugars explains its difference in kinetic and biophysical properties of its isoforms [64][52].
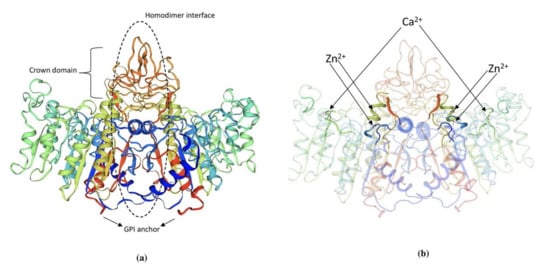
Figure 2. (a) 3D structure of human PLAP indicating crown domain, GPI anchor and the homodimer interface regions. (b) The presence of zinc and calcium ions is clearly indicated in the PLAP structure. The structure was exported from https://swissmodel.expasy.org/, accessed on 1 August 2021. (P05186 (PPBT_HUMAN)).
2.3. Mechanism of Mineralization-Promotion and Inhibition
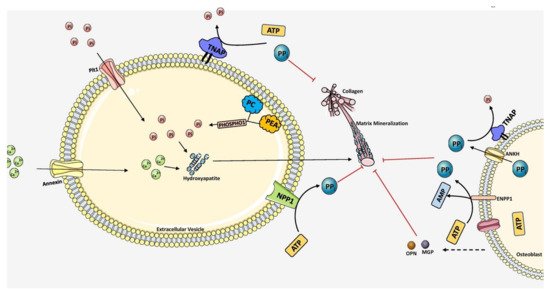
Biomineralization diversifies the mechanical properties of various connective tissues including bone. For instance, mineralization is absent in type 1 collagen of dermis to maintain skin texture, while it is highly mineralized in bone and teeth to form rigid structures [65][53]. Mineralization of bone follows a two-step process. Initial step involves the formation of HAP inside the EVs and followed by the propagation into the extracellular matrix. The EVs are typically 50-200 nm in diameter which are produced from the plasma membrane of osteoblasts, chondroblasts and odontoblasts [33][20]. The mechanism behind the release of EVs forms these cells is not yet known. Interestingly, the EVs differ in their membrane composition compared to the originating cell type, marked with the presence of several phospholipids, specifically phosphatidylserine which shows high affinity for calcium ions [66,67,68,69][54][55][56][57]. EVs are also rich in annexins A5, A2, A6 and calbindin D9k, ALP, carbonic anhydrase, collagen x, type III sodium-phosphate transporter, Phosphatase, Orphan 1 (PHOSPHO1), and nucleotide pyrophosphate phosphodiesterase [70,71,72,73][58][59][60][61]. Calcium binding proteins, phospholipids, and bone sialoprotein (BSP) mediate calcium accumulation in EVs [74][62]. Incorporation of calcium in EVs is facilitated by the formation of calcium channels by membrane bound annexins. The role of various annexins and matrix vesicles in bone mineralization is discussed in depth and more insights can be acquired by reading Ansari et al. [75][63]. Next, phosphates are provided by type III Na/Pi cotransporter present on both EVs membrane and cell membrane [76,77][64][65]. In addition to this, PHOSPHO1, a cytosolic phosphatase also contributes to phosphate levels by catalyzing the hydrolysis of phosphocholine and phosphoethanolamine [78,79][66][67]. When the accumulation of both calcium and phosphate exceeds solubility point for calcium phosphate it leads to the deposition as hydroxyapatite inside the EVs. In the next step of mineralization, hydroxyapatite crystals penetrate EVs membrane to reach the extracellular space and undergo crystal elongation. Elongation is highly dependent on extracellular concentrations of calcium and phosphate ions outside the EVs [73,75][61][63]. Sufficient levels of calcium and Pi support the formation of new apatite crystals, which propagate in clusters around the EVs and fill in between collagen fibrils in the ECM in a cross-fibrillar pattern [19]. The mechanism of mineralization is diagrammatically represented in Figure 3.
Figure 3. Overall scheme of matrix mineralization mediated by extracellular vesicles (EV) released from osteoblasts. The growth of hydroxyapatite inside EVs is mediated by the PHOSPHO1 which supplies Pi by hydrolyzing PC and PEA in the plasma membrane. Pit1 transporter also pumps Pi into the EVs. Calcium is transported inside by Annexin (A1, A2, A4, A5, A6, A7) channels. Together, hydroxyapatite crystals are formed inside the EV. It then penetrates through the vesicle membrane and elongates extracellularly utilizing Pi and Ca2+ ions. PPi inhibits mineralization which is produced by ATP hydrolysis mediated by NPP1. TNAP hydrolyzes PPi into Pi, essential for hydroxyapatite growth. Extracellular PPi is also provided by ANKH and ENPP1 situated in the plasma membrane of osteoblasts. Mineralization is regulated tightly by maintaining the balance between PPi and Pi ratio. The components in this figure were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. (http://smart.servier.com, accessed on 1 August 2021).
Propagation around clusters is mediated cooperatively by EVs and adjacent collagen molecules. Since mineralization is central to bone, our body prevents ectopic biomineralization by various ingenious inhibitors. In bone, to eliminate the inhibitors of biomineralization especially PPi, osteoblasts express TNAP to annul its inhibitory effect via hydrolysis and generate phosphate ions promoting mineralization. TNAP not only provides phosphate ions for mineralization it also promotes the formation of HAP nanocrystals in the collagen matrix of bone by destabilizing the amorphous phase of the mineral. Also, local inhibitors of biomineralization attenuates pathological mineralization in soft tissues such as cardiac arteries, valves and hard/soft tissue interfaces such as tendon-bone attachments, ligament-bone attachments and cranial sutures. It is noteworthy to mention that, the inorganic phosphate (Pi) to pyrophosphate (PPi) ratio is crucial for mineralization as PPi inhibits hydroxyapatite formation [80][68]. ANKH, a human homolog of ank gene product, is essential for the extracellular transportation of cytosolic PPi [81,82][69][70]. An increase in PPi/Pi ratio will inhibit spontaneous precipitation of HAP. The PPi concentration is rightly maintained in forming bone by osteoblasts and acts as a repository of compartmentalized phosphate till it is hydrolyzed [80][68]. Extracellular polyphosphate (polyPi) also participates in phosphates transport and compartmentalization. Poly-Pi granules chelate calcium ions to form neutrally charged amorphous complexes [83,84][71][72]. Fetuin A is a systemic inhibitor of mineralization which prevents growth of nascent crystal nuclei in blood and facilitates its recycling by macrophages [85,86,87,88][73][74][75][76]. One fetuin is known to sequester 54–72 phosphate ions and 90–120 calcium ions. Fetuin A possesses strong affinity to bone and constitutes 25% of the non-collagenous proteins in bone [89][77]. It binds with both Ca and PO42− ions to form calciprotein particles about 30–150 nm in size [90][78]. TNAP, expressed highly in osteoblasts, is very essential for the second step of mineralization by decreasing the levels of PPi and providing Pi for hydroxyapatite formation. Therefore, in bone, mineralization is a well-coordinated, cell mediated dynamic process allowing the exchange of phosphate and calcium ions. In soft tissues, mineralization is generally inhibited by high PPi/Pi ratio. The polyPi granules are also known to contain alkaline phosphatases and when activated, hydroxyapatite crystals nucleate inside the granules displacing protein components to the surface forming a crystalline core surrounded by an amorphous shell [84][72]. Skeletal tissue biomineralization is also promoted by bone sialoprotein (BSP), i.e., a calcium-binding small integrin-binding ligand N-linked glycoprotein (SIBLING) aids in nucleation of hydroxyapatite mineral [91][79]. It is only present in the ECM of mineralized tissues such as bone and teeth. Another promoter of ECM mineralization, Dentin matrix acidic phosphoprotein1 (DMP1) is involved in the stabilization of disordered mineral precursors and directs them to collagen fibrils [92,93][80][81]. DMP1 binds with mineral forming mineral-protein complexes facilitating electrostatic interaction with collagen fibrils during mineralization. Patients with DMP1 gene deletions or mutations are characterized by osteomalacia with autosomal recessive hypophosphatemic rickets [94][82]. OPN and matrix Gla protein (MGP) are mineralization inhibiting proteins which regulate mineral growth in bone and dentition to adjust mineralization or involve complete inhibition of mineralization [95,96][83][84]. Nascent mineralization foci are associated with osteopontin (OPN) and found widely in the bone matrix. For instance, OPN deficient mice exhibit a hypermineralized skeleton postnatally starting at 12 weeks [97][85]. OPN plays a crucial role as interfacial protein in adjoining new bone to old bone and also to implant surfaces following bone graft surgeries. It is actively found in sites where mineralization should be abolished abruptly such as periodontal ligament, and entheses [98][86]. OPN knock-out mice failed to curb crystal growth with increased size of mineral crystals but do not show any visible skeletal changes [99][87].
Despite the precise inhibition of mineralization at soft-tissue sites, arteries and articular cartilage are susceptible for ectopic mineralization which leads to detrimental effects. Therefore, mineralization inhibition requires strict regulation involving additional elements. Vascular smooth muscle cells and chondrocytes orchestrate this inhibition by secreting Matrix Gla protein (MGP) into their extracellular matrix (ECM) [100][88]. Singleton Merten syndrome and Keutel syndrome associated with defects in MGP gene results in rupture of the aorta and premature fusion of growth plates in the long bone due to pathological mineralization [101][89]. Other mineralization inhibitors such as aspartic acid-rich motif (ASARM) and matrix extracellular phosphoglycoprotein (MEPE) also fine tune mineralization [102,103][90][91]. MEPE knock out mice show increased bone mass and trabecular density but abnormalities in cancellous bone [104][92]. MEPE along with dentin matrix protein (DMP1) and Phosphate-regulating neutral endopeptidase (PHEX) regulates mineralization, phosphate levels and turnover of bone by affecting fibroblast growth factor (FGF)-23 expression [105][93]. Imbalance in mineralization promotion and inhibition at required areas in the body leads to various pathologies. The following sections, we will discuss the pivotal role of TNAP in various organs with respect to mineralization.
2.4. Hypophosphatasia-Physiological Substrates of TNAP
In humans, endochondral ossification and intramembranous ossification is dependent on TNAP activity. A common manifestation of loss of TNAP activity or mutations to ALPL gene causes a systemic bone disease called hypophosphatasia (HPP) resulting in hypomineralization of hard skeletal tissues including bone and teeth [106,107][94][95]. Several missense mutations are reported in human ALPL gene corresponding to aminoacid changes in the protein [106,107,108][94][95][96]. At the time of writing this review, 409 mutations in TNAP have been reported owing to its clinical heterogeneity. The mutant form of TNAP possesses variable kinetic properties for PLP and PPi [109][97]. The clinical expression of this disease is highly variable affecting patients of all ages. It is a multisystemic disease affecting various tissues such as bone, kidney, muscle, GI tract, lung and central nervous system (CNS). Milder forms of the disease include deformities of extremities, early deciduous teeth loss and other dental manifestations. In severe cases of HPP, prenatal death of foetuses is seen due to complete absence of minerals in their skeleton [106,107,108][94][95][96]. Epileptic seizures, craniosynostosis, respiratory failure and death are also associated in extreme forms of HPP. As far as treatment regime is concerned, there are no curative treatment options available for HPP and it is treated symptomatically requiring a multidisciplinary team and approach [110][98]. Enzyme replacement therapy (ERT) includes Human recombinant enzyme Asfotase alfa which is approved for patients with pediatric onset of HPP to treat various skeletal and respiratory manifestations (NCT02797821). ERT improves growth, motor function, agility, strength and reduced pain in children with HPP [111][99]. Patients with a severe life-threatening form of HPP also showed substantial improvement in bone mineralization and survival. In humans and mice models of HPP, the TNAP-deficient EVs’ extracellular growth of HA crystals is blocked by excessive extracellular accumulation of PPi [112][100]. Combined ablation of PHOSPHO1 and TNAP results in the absence of HA crystals within EVs, absence of skeletal mineralization and embryonic deadliness [113][101]. Between TNAP and PHOSPHO1 exists a cross talk in the mineralization initiation [114][102]. PHOSPHO1 is essential for Pi generation within MVs which is necessary for the initiation of HA formation inside the vesicle. TNAP is a crucial PPiase essential for the extracellular growth of HA by supplying Pi via PPi hydrolysis [112][100]. Ecto-Nucleotide Pyrophosphatase/ Phosphodiesterase-1 (NPP1), a cell surface enzyme on EVs is a potent ATPase which produces PPi and acts as a phosphatase in the absence of TNAP [115][103]. NPP1 may modify HPP phenotype in experimental models. For instance, in PHOSPHO1 deficient mice, increase PPi levels was detected in plasma caused by reciprocal decrease in TNAP activity and elevated NPP1 expression. Interestingly, pOPN circulatory levels were increased, an inhibitor of mineralization which furthers the risk of fracturing in HPP [50][37]. The convergent step involving influx of Pi produced by NPP1 and TNAP and production of Pi by PHOSPHO1 inside the EVs suggests the combined interactions between TNAP, NPP1 and PHOSPHO1 during EVs mediated calcification [50][37].
By investigating HPP patients, PLP, which is a circulating form of vitamin B6, was found to be another natural substrate of TNAP. PLP is coenzyme essential for various neurotransmitter’s synthesis [116][104] and its entry into neuronal cells is promoted by removal of Pi mediated by TNAP [22][105]. Increased plasma PLP levels were seen in HPP patients with TNAP deficiency. In the severe form of HPP, infants display vitamin B6-dependent seizures resulting from low circulatory levels of PL and inefficient incorporation into CNS [117,118][106][107]. The importance of TNAP in CNS is shown by its presence in developing murine neural tube and areas of the mature brain [119,120][108][109]. Increased amounts of less developed cortical synapses, hypomyelination and spinal nerves thinning are seen in TNAP KO mice [41,42,121][28][29][110]. Ingestion of PL temporarily suppressed epilepsy in TNAP KO mice. This evidence suggests the role of TNAP in both developing and mature CNS.
Another biological substrate of TNAP is PEA, whose level was elevated in urine and blood of HPP patients [122][111]. PEA is a component of glycosylphosphatidylinositol link for proteins like TNAP. TNAP is essential for the extracellular hydrolysis of PLP to PL and transport into cells for PLP formation, a vital cofactor in many reactions. Hepatic O-phosphorylethanolamine phospholyase (PEA-P-lyase) enzyme is reported to be involved in PEA hydrolysis using PLP as cofactor [123][112]. In HPP patients, insufficiency in the intracellular levels of PLP in hepatic cells could be the reason for elevated PEA accumulation and its rise in blood and urine levels. Despite this, the exact mechanism is yet to be known behind this PEA accumulation in HPP patients.
Yet another pathogenicity in HPP is increased plasma OPN (encoded by Spp1) levels suggesting that it may be another potential substrate of TNAP. Phosphorylation of OPN inhibits mineral deposition [124][113]. However, the exact role of OPN is not completely understood. OPN is known to anchor osteoclasts to HA minerals by poly-aspartate sequences and bind CD44 and αvβ3 integrin with RGD, which mediates cell migration and cell signaling [125][114]. Phosphorylated form of OPN inhibits mineralization in vascular smooth muscle cells [126][115]. High levels of phosphorylated OPN and extracellular PPi levels were observed in Alpl KO mice. Double knockout of Alpl−/− and Spp1−/− in mice models partially improved hypomineralization compared to Alp−/− KO mice [127][116]. Therefore, TNAP loss leads to accumulation of phosphorylated OPN which in turn results in impaired bone mineralization in murine HPP. Similarly, PHOSPHO1 deficient mice models exhibited an increase in levels of both PPi and phosphorylated OPN. Ablation of Spp1 reverts the skeletal deficiency in Phospho1 deficient mice [50][37]. Altogether, it is clear that TNAP exhibits a wide range of substrates and whose levels are disrupted in TNAP dysfunction.
2.5. Role of TNAP as An Anti-Inflammatory Enzyme
TNAP is available as a soluble isoform in blood primarily originating from bone and liver tissue. Bone is the main source of TNAP during skeletal growth and it slowly progresses down with the aging process, thus the liver becomes the major source of TNAP in blood. It is very challenging to distinguish between bone and liver TNAPs in blood as bone TNAP exhibits 18% cross reactivity with liver TNAP [128,129][117][118]. Circulating TNAP exert an anti-inflammatory role by contributing to the generation of adenosine from AMP, detoxification by LPS dephosphorylation and regulating postprandial endotoxaemia [130,131][119][120]. In the HPP model with TNAP deficiency, bone marrow edema [132][121], osteomyelitis [133][122], tendinitis [134][123] and increased predisposition to periodontitis is widely seen among both children and adults [135][124]. TNAP through its ectonucleotidase activity exerts a major role in balancing pro-inflammatory ATP levels and anti-inflammatory role through adenosine, a breakdown product of ATP [136][125]. Owing to this, it has gained much attention leading to various studies investigating several agonists and antagonists. Impairment in TNAP ectophosphatase activity leads to PPi accumulation initiating the formation of calcium pyrophosphate dihydrate crystals [137][126]. These crystals which accumulate in tissue and joints of HPP patients trigger necroinflammation and NLRP3 inflammasome activation [138][127]. OPN is a natural substrate of TNAP is thought to be involved in mineralization. However, it is a potent proinflammatory protein and status of pro- and anti-inflammatory properties are attributed to the status of its phosphorylation and dephosphorylation mediated by TNAP. Very recently, recombinant OPN was found to mediate anti-inflammatory cascades in microglia of the brain by inhibition of NLRP3 inflammasome [139][128]. OPN also regulates inflammatory processes in kidney and liver. However, in-depth investigation dissecting the role of TNAP mediated OPN dephosphorylation in balancing pro- and anti-inflammatory effects is necessary. The ectophosphatase activity of TNAP is also associated with the dephosphorylation of TLR ligands, such as double-stranded RNA; mimic poly-inosine:cytosine, a TLR3 agonist; and microbial LPS, a TLR4 ligand that mitigates the activation of inflammasome and the secretion of cytokines in sepsis [131][120]. TNAP is also known to modulate T-cell function in a preclinical model of intestinal colitis [140][129]. TNAP balances P1 and P2 receptor mediated signaling in modulating the level of the inflammatory process. TNAP present in the neutrophil cell membrane hydrolyze AMP, LPS and PLP and controls autocrine effects of adenosine on neutrophil migration, survival and IL-1α secretion [136,141,142][125][130][131]. TNAP in the membrane of endothelial cells may be involved in LPS dephosphorylation [131][120]. In 7-day old Alpl+/− mice bone metaphysis, the levels of IL-1α and IL-6 were increased and IL-10 anti-inflammatory levels were decreased compared with Alpl+/+ mice. In hypertrophic chondrocytes, TNAP inhibition had no effect on ATP and adenosine associated changes including the modulation of autocrine pro-inflammatory effects. In neutrophils, inhibition of TNAP worsened ATP induced secretion of IL-1α and reduced cell survival [136][125]. AP infusion in several phase 2 clinical trials has shown improvement in the outcome of various inflammatory diseases such as sepsis, inflammatory bowel disease and ischemia/reperfusion [143][132]. Overall, the role of TNAP in modulating inflammation and further investigations are needed to decipher the exact mechanisms behind its mode of action.
2.6. Role of TNAP in Central Nervous System
TNAP is also expressed by neuronal and endothelial cells where it plays a crucial role in brain development. Interestingly, TNAP is very active in the arterial part but not at the venal part of the microvasculature [144,145][133][134]. It can be detected in human brain blood vessels right from gestational ages and this distribution of TNAP hints its involvement in active transport of molecules across the blood brain barrier [146,147][135][136]. It is very active in both luminal as well as abluminal sides of the endothelial cell membrane mediating transport across these cells. The possible role of TNAP includes PLP dephosphorylation and pyridoxal transport across epithelial cells of capillary. Epileptic seizures, decreased GABA production and elevated levels of PLP in blood of Alpl-deficient mice with HPP dictates the vital role of TNAP in the nervous system. Apart from endothelial TNAP, its presence in neurons is very important in normal brain development as it is widely seen in both white and grey matter during developmental stages of the brain [148][137]. Surprisingly, elevated TNAP activity is also observed in adult neurogenesis. Delayed myelination and spinal abnormalities are seen in Alpl-deficient mice. MRI investigations of HPP infants shows hypodensity of white matter, multicystic encephalopathy, dilated ventricles, and parenchymal lesions [149,150][138][139]. Dephosphorylation and interaction with ECM proteins including collagen and laminin is also contributed by TNAP in brain development [151][140]. Post mortem of the brain from patients with Alzheimer’s disease (AD) showed increased TNAP protein levels and its activity in temporal gyrus and hippocampus, regions targeted by tau protein accumulation [152,153][141][142]. TNAP may exhibit dual effects in AD. First, it ameliorates neuroinflammation by adenosine synthesis through ATP dephosphorylation and maintains a normal functional BBB [154][143]. In contrast, it participates in dephosphorylation of hyperphorylated extracellular Tau protein which interacts with muscarinic receptors disrupting calcium homeostasis which leads to neuronal death [153][142]. TNAP inhibition may mediate brain damage induced by ischemic stroke events [155][144]. Therefore, it is regarded as an important biomarker and mediates events following ischemic reperfusion injuries. Patients with acute ischemic stroke show an elevated level of TNAP in serum which is associated with stroke recurrence and death [156,157,158,159][145][146][147][148]. On the other hand, TNAP may also attenuate neuroinflammation after stroke by dephosphorylation of ATP which is released in large amounts following cell necrosis [160][149]. Thus, TNAP blockade may lead to worsening of brain damage and contribute to neuroinflammation and therefore, interventions on TNAP inhibition require a thorough investigation.
2.7. Hepatic Role of TNAP
Although the expression of TNAP is known, its function remains obscure in the kidney and liver. In hepatocytes, TNAP is located at the canalicular membrane and in the apical area in the cytoplasm, in bile duct of epithelial cells [161,162][150][151]. It is involved in bile excretion and participates in bile pH regulation by dephosphorylating ATP at the surface of cholangiocytes [163][152]. Circulatory levels of liver TNAP is largely seen in cholangitis suggesting its role in bile excretion. In addition to this, it may involve detoxification and biliary excretion of LPS [164][153]. During systemic inflammation, in adults, TNAP from the liver is largely released into the blood when bone formation is compromised and bone TNAP levels are reduced in blood. However, the exact function of hepatic TNAP is still unknown.
2.8. Renal Role of TNAP
In the kidneys, proximal renal tubules’ brush border expresses TNAP and is involved in LPS detoxification by dephosphorylation [164,165][153][154]. Renal tissue express TNAP, an activator of mineralization. One would expect mineralization in renal tissue due to the presence of TNAP but interestingly, urine mineralization is prevented by PPi. The PPi production site is at the distal nephron; there is no TNAP expression there, which explains the inability of TNAP in the renal system to trigger urinary tract calcification [166][155]. TNAP inhibitors blocked norepinephrine dependent renovascular and blood pressure responses indicating the importance of TNAP in the kidney. The detrimental role of TNAP is seen in chronic kidney disease (CKD) where its activation leads to arterial media calcification resulting in stiffening of the blood vessels [167,168][156][157]. In CKD, calcification inhibitors such as MGP and PPi are decreased which results in ectopic mineralization of the artery wall [169][158]. The precise balance between PPi/Pi is perturbed leads to hydroxyapatite mineralization. Inhibition of TNAP activity leads to an improvement in CKD pathologies. Additional information on vascular calcification is discussed below.
2.9. TNAP in Vascular Calcification
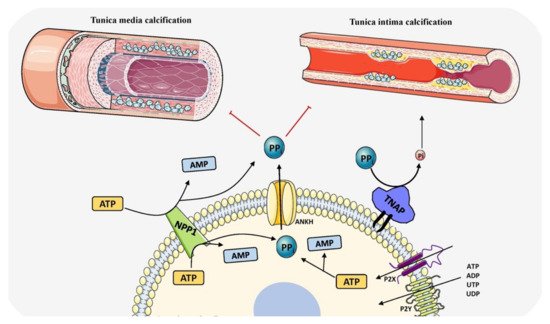
Cardiovascular calcification (CVC) is an important risk factor for morbidity and mortality in patients of all ethnicities and increases with age [170][159]. Age related calcification includes aortic valve calcification and atherosclerotic plaque calcification and in chronic kidney disease and type 2 diabetes, tunica media calcification is largely seen to be associated with increased mortality risk [171][160]. Cardiovascular mortality risk is positively linked to calcium levels [172][161]. Until recently, TNAP was found to be a central player in CVC and opened new avenues for treating and managing the disease. All CVC types mimic either endochondral or intramembranous calcification and both have been seen in the arterial tunica media of both rodents and humans with CKD and diabetes (Figure 4). Both physiological and pathological calcification share the same molecular events leading to ECM mineralization.
Figure 4. Schematic representation of the role of TNAP in vascular calcification by determining PPi/Pi ratio and balancing induction and inhibition of mineralization. The components in this figure were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License (http://smart.servier.com, accessed on 1 August 2021).
Calcifying vascular specimens shows the presence of osteoblasts, osteoclasts and chondrocytes which were derived from stem cells or by transdifferentiation of vascular smooth muscle cells [173,174,175,176][162][163][164][165]. Local TNAP activation triggers massive vascular calcification in arteries owing to its overexpression in vascular smooth muscle cells and endothelial cells [177,178][166][167]. Likewise, circulating TNAP is also associated with increased mortality in patients with CKD where the serum TNAP is found to be elevated. In patients undergoing hemodialysis, plasma pyrophosphate levels are reduced following dialysis and it is associated with aortic wall vascular calcifications [179,180][168][169]. Involvement of the TNAP/PPi system is interesting and is worth investigating to subdue and inhibit VC. Both TNAP and NPP1 enzymes are present on the cell membrane of calcifying cells to precisely regulate levels of PPi, inhibitor of mineralization by blocking the expansion of nascent hydroxyapatite crystals. Routine administration of exogenous pyrophosphate prevented vascular calcification (VC) in mice and rat models [181,182][170][171]. In mice models with ank mutation or NPP1-/- exhibit vascular changes through the alteration of cartilage specific genes. In humans, NPP1 deficiency causes idiopathic infantile arterial calcification which affects children resulting in medial calcification of arteries [183,184][172][173]. Early microcalfications is targeted by bisphosphonates (non-hydrolysable PPi analogues) preventing vessel wall mineralization. TNAP activity is more prominently observed in uremic rats and mouse models of medial calcification [185][174]. The dual functions of ALP well known to release Pi by phosphomonoesters hydrolysis and act as pyrophosphatases in bone. However, its function outside bone is uncertain. In HPP patients, with elevated TNAP activity there is no clear evidence of vascular calcification indicating that only membrane bound TNAP and other cofactors are required to induce VC. Finally, transdifferentiation of SMCs to chondrocytes via BMP-2 activation and calcium deposition is stimulated by TNAP which explains the role in VC [186][175]. Overexpression of TNAP induces medial vascular calcification in an ex vivo model of rat aortic rings [9,187][9][176]. Although pharmacological inhibition of ALP and PHOSPHO1 also suppressed vascular smooth muscle cell calcification, it induced loss of skeletal mineralization [188,189][177][178]. Despite various reports, it is very hard to conclude the involvement of amplified TNAP levels in vascular calcification. Greater understanding of the mechanisms involving TNAP in vascular calcification is needed to better study the clinical consequences and develop therapeutic agents.
References
- Kenkre, J.S.; Bassett, J.H.D. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327.
- Kirsch, T. Determinants of pathological mineralization. Curr. Opin. Rheumatol. 2006, 18, 174–180.
- Kirsch, T. Physiological and pathological mineralization: A complex multifactorial process. Curr. Opin. Orthop. 2007, 18, 425–427.
- Villa-Bellosta, R.; Egido, J. Phosphate, pyrophosphate, and vascular calcification: A question of balance. Eur. Heart J. 2015, 38, ehv605-1804.
- Blumenthal, H.T.; Lansing, A.I.; Wheeler, P.A. Calcification of the Media of the Human Aorta and Its Relation to Intimal Arteriosclerosis, Ageing and Disease. Am. J. Pathol. 1944, 20, 665–687.
- Nicoll, R.; Henein, M.Y. The predictive value of arterial and valvular calcification for mortality and cardiovascular events. IJC Heart Vessel. 2014, 3, 1–5.
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc. Dis. 2012, 1, 1–10.
- Shanahan, C.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711.
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells during Phosphate-Induced Calcification. Arter. Thromb. Vasc. Biol. 2018, 38, 2137–2147.
- Yamato, Y.; Matsukawa, M.; Mizukawa, H.; Yanagitani, T.; Yamazaki, K.; Nagano, A. Distribution of hydroxyapatite crystallite orientation and ultrasonic wave velocity in ring-shaped cortical bone of bovine femur. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2008, 55, 1298–1303.
- Osaki, S.; Tohno, S.; Tohno, Y.; Ohuchi, K.; Takakura, Y. Determination of the orientation of collagen fibers in human bone. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2002, 266, 103–107.
- Glimcher, M.J.; Muir, H. Recent studies of the mineral phase in bone and its possible linkage to the organic matrix by protein-bound phosphate bonds. Philos. Trans. R. Soc. B Biol. Sci. 1984, 304, 479–508.
- Landis, W.; Song, M.; Leith, A.; McEwen, L.; McEwen, B.F. Mineral and Organic Matrix Interaction in Normally Calcifying Tendon Visualized in Three Dimensions by High-Voltage Electron Microscopic Tomography and Graphic Image Reconstruction. J. Struct. Biol. 1993, 110, 39–54.
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; De With, G.; Sommerdijk, N.A.J.M. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010, 9, 1004–1009.
- Traub, W.; Arad, T.; Weiner, S. Origin of Mineral Crystal Growth in Collagen Fibrils. Matrix 1992, 12, 251–255.
- Anderson, H.C. The role of matrix vesicles in physiological and pathological calcification. Curr. Opin. Orthop. 2007, 18, 428–433.
- Van de Lest, C.H.A.; Vaandrager, A.B. Mechanism of cell-mediated mineralization. Curr. Opin. Orthop. 2007, 8, 434–443.
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene 2020, 754, 144855.
- Nair, A.; Gautieri, A.; Chang, S.-W.; Buehler, M.J. Molecular mechanics of mineralized collagen fibrils in bone. Nat. Commun. 2013, 4, 1724.
- Orimo, H. The Mechanism of Mineralization and the Role of Alkaline Phosphatase in Health and Disease. J. Nippon. Med. Sch. 2010, 77, 4–12.
- Weiss, M.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010.
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millan, J.L.; MacGregor, G.R.; et al. Alkaline Phosphatase Knock-Out Mice Recapitulate the Metabolic and Skeletal Defects of Infantile Hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026.
- Yadav, M.K.; Manoli, N.M.; Vimalraj, S.; Madhunapantula, S.V. Unmethylated promoter DNA correlates with p53 expression and apoptotic levels only in Vitamin B9 and B12 deficient megaloblastic anemia but not in non-megaloblastic anemia controls. Int. J. Biol. Macromol. 2018, 109, 76–84.
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010, 1192, 190–200.
- Hessle, L.; Johnson, K.; Anderson, H.C.; Narisawa, S.; Sali, A.; Goding, J.W.; Terkeltaub, R.; Millán, J.L. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. USA 2002, 99, 9445–9449.
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank: An Integrated Model of the Pathogenesis of Mineralization Disorders. Am. J. Pathol. 2004, 164, 1199–1209.
- Amadasi, A.; Bertoldi, M.; Contestabile, R.; Bettati, S.; Cellini, B.; Di Salvo, M.L.; Borri-Voltattorni, C.; Bossa, F.; Mozzarelli, A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324.
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, S.R. Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51.
- Narisawa, S.; Wennberg, C.; Millán, J.L. Abnormal vitamin B6 metabolism in alkaline phosphatase knock-out mice causes multiple abnormalities, but not the impaired bone mineralization. J. Pathol. 2000, 193, 125–133.
- Michigami, T.; Ohata, Y.; Fujiwara, M.; Mochizuki, H.; Adachi, M.; Kitaoka, T.; Kubota, T.; Sawai, H.; Namba, N.; Hasegawa, K.; et al. Clinical Practice Guidelines for Hypophosphatasia. Clin. Pediatr. Endocrinol. 2020, 29, 9–24.
- Lei, W.; Ni, H.; Herington, J.; Reese, J.; Paria, B.C. Alkaline Phosphatase Protects Lipopolysaccharide-Induced Early Pregnancy Defects in Mice. PLoS ONE 2015, 10, e0123243.
- Yang, Y.; Wandler, A.M.; Postlethwait, J.H.; Guillemin, K. Dynamic Evolution of the LPS-Detoxifying Enzyme Intestinal Alkaline Phosphatase in Zebrafish and Other Vertebrates. Front. Immunol. 2012, 3, 314.
- Sebastián-Serrano, Á.; de Diego-García, L.; Henshall, D.C.; Engel, T.; Diaz-Hernandez, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9, 170.
- Woods, P.S.; Doolittle, L.M.; Hickman-Davis, J.M.; Davis, I.C. ATP catabolism by tissue nonspecific alkaline phosphatase contributes to development of ARDS in influenza-infected mice. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L83–L92.
- Zhang, Z.; Nam, H.; Crouch, S.; Hatch, N. Tissue Nonspecific Alkaline Phosphatase Function in Bone and Muscle Progenitor Cells: Control of Mitochondrial Respiration and ATP Production. Int. J. Mol. Sci. 2021, 22, 1140.
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In Vivo Overexpression of Tissue-Nonspecific Alkaline Phosphatase Increases Skeletal Mineralization and Affects the Phosphorylation Status of Osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598.
- Yadav, M.C.; Huesa, C.; Narisawa, S.; Hoylaerts, M.F.; Moreau, A.; Farquharson, C.; Millán, J.L. Ablation of Osteopontin Improves the Skeletal Phenotype of Phospho1−/− Mice. J. Bone Miner. Res. 2014, 29, 2369–2381.
- Addison, W.N.; Azari, F.; Sørensen, E.S.; Kaartinen, M.T.; McKee, M.D. Pyrophosphate Inhibits Mineralization of Osteoblast Cultures by Binding to Mineral, Up-regulating Osteopontin, and Inhibiting Alkaline Phosphatase Activity. J. Biol. Chem. 2007, 282, 15872–15883.
- Weiss, M.; Henthorn, P.S.; Lafferty, M.A.; Slaughter, C.; Raducha, M.; Harris, H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 7182–7186.
- Kiledjian, M.; Kadesch, T. Analysis of the human liver/bone/kidney alkaline phosphatase promoter In Vivo and In Vitro. Nucleic Acids Res. 1990, 18, 957–961.
- Orimo, H.; Shimada, T. Regulation of the human tissue-nonspecific alkaline phosphatase gene expression by all-trans-retinoic acid in SaOS-2 osteosarcoma cell line. Bone 2005, 36, 866–876.
- Orimo, H.; Shimada, T. Posttranscriptional modulation of the human tissue–nonspecific alkaline phosphatase gene expression by 1,25-dihydroxyvitamin D3 in MG-63 osteoblastic osteosarcoma cells. Nutr. Res. 2006, 26, 227–234.
- Orimo, H.; Shimada, T. The role of tissue-nonspecific alkaline phosphatase in the phosphate-induced activation of alkaline phosphatase and mineralization in SaOS-2 human osteoblast-like cells. Mol. Cell. Biochem. 2008, 315, 51–60.
- Delgado-Calle, J.; Sañudo, C.; Sánchez-Verde, L.; García-Renedo, R.J.; Arozamena, J.; Riancho, J.A. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone 2011, 49, 830–838.
- Henthom, P.S.; Raducha, M.; Hadesch, T.; Weiss, M.J.; Harris, H. Sequence and characterization of the human intestinal alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12011–12019.
- Knoll, B.J.; Rothblum, K.N.; Longley, M. Nucleotide sequence of the human placental alkaline phosphatase gene. Evolution of the 5′ flanking region by deletion/substitution. J. Biol. Chem. 1988, 263, 12020–12027.
- Griffin, C.A.; Smith, M.; Henthorn, P.S.; Harris, H.; Weiss, M.; Raducha, M.; Emanuel, B.S. Human placental and intestinal alkaline phosphatase genes map to 2q34-q37. Am. J. Hum. Genet. 1987, 41, 1025–1034.
- Millán, J.L. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology; Wiley-VCH GmbH & Co.: Weinheim, Germany, 2006.
- Pike, A.F.; Kramer, N.I.; Blaauboer, B.J.; Seinen, W.; Brands, R. A novel hypothesis for an alkaline phosphatase ‘rescue’ mechanism in the hepatic acute phase immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2044–2056.
- Silvent, J.; Gasse, B.; Mornet, E.; Sire, J.-Y. Molecular Evolution of the Tissue-nonspecific Alkaline Phosphatase Allows Prediction and Validation of Missense Mutations Responsible for Hypophosphatasia. J. Biol. Chem. 2014, 289, 24168–24179.
- Mornet, E.; Stura, E.; Lia-Baldini, A.-S.; Stigbrand, T.; Ménez, A.; Le Du, M.-H. Structural Evidence for a Functional Role of Human Tissue Nonspecific Alkaline Phosphatase in Bone Mineralization. J. Biol. Chem. 2001, 276, 31171–31178.
- Nosjean, O.; Koyama, I.; Goseki, M.; Roux, B.; Komoda, T. Human tissue non-specific alkaline phosphatases: Sugar-moiety-induced enzymic and antigenic modulations and genetic aspects. Biochem. J. 1997, 321, 297–303.
- Reznikov, N.; Steele, J.; Fratzl, P.; Stevens, M.M. A materials science vision of extracellular matrix mineralization. Nat. Rev. Mater. 2016, 1, 16041.
- Davies, O.G.; Cox, S.C.; Azoidis, I.; McGuinness, A.; Cooke, M.; Heaney, L.; Davis, E.T.; Jones, S.W.; Grover, L.M. Osteoblast-Derived Vesicle Protein Content Is Temporally Regulated during Osteogenesis: Implications for Regenerative Therapies. Front. Bioeng. Biotechnol. 2019, 7, 92.
- Masaoutis, C.; Theocharis, S. The Role of Exosomes in Bone Remodeling: Implications for Bone Physiology and Disease. Dis. Markers 2019, 2019, 9417914.
- Skotland, T.; Sagini, K.; Sandvig, K.; Llorente, A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 2020, 159, 308–321.
- Wuthier, R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta Lipids Lipid Metab. 1975, 409, 128–143.
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular Vesicles as Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 2017, 4, 78.
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional Involvement of PHOSPHO1 in Matrix Vesicle-Mediated Skeletal Mineralization. J. Bone Miner. Res. 2007, 22, 617–627.
- Solomon, D.H.; Wilkins, R.J.; Meredith, D.; Browning, J.A. Characterisation of Inorganic Phosphate Transport in Bovine Articular Chondrocytes. Cell. Physiol. Biochem. 2007, 20, 099–108.
- Golub, E.E. Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1592–1598.
- Nahar, N.N.; Missana, L.; Garimella, R.; Tague, S.E.; Anderson, H.C. Matrix vesicles are carriers of bone morphogenetic proteins (BMPs), vascular endothelial growth factor (VEGF), and noncollagenous matrix proteins. J. Bone Miner. Metab. 2008, 26, 514–519.
- Ansari, S.; de Wildt, B.; Vis, M.; de Korte, C.; Ito, K.; Hofmann, S.; Yuana, Y. Matrix Vesicles: Role in Bone Mineralization and Potential Use as Therapeutics. Pharmaceuticals 2021, 14, 289.
- Lundquist, P.; Murer, H.; Biber, J. Type II Na+-Pi Cotransporters in Osteoblast Mineral Formation: Regulation by Inorganic Phosphate. Cell. Physiol. Biochem. 2007, 19, 43–56.
- Yadav, M.C.; Bottini, M.; Cory, E.; Bhattacharya, K.; Kuss, P.; Narisawa, S.; Sah, R.L.; Beck, L.; Fadeel, B.; Farquharson, C.; et al. Skeletal Mineralization Deficits and Impaired Biogenesis and Function of Chondrocyte-Derived Matrix Vesicles in Phospho1−/− and Phospho1/Pit1 Double-Knockout Mice. J. Bone Miner. Res. 2016, 31, 1275–1286.
- Roberts, S.J.; Stewart, A.J.; Sadler, P.J.; Farquharson, C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem. J. 2004, 382, 59–65.
- Stewart, A.J.; Leong, D.T.K.; Farquharson, C. PLA 2 and ENPP6 may act in concert to generate phosphocholine from the matrix vesicle membrane during skeletal mineralization. FASEB J. 2018, 32, 20–25.
- Fleisch, H.; Russell, R.G.G.; Straumann, F. Effect of Pyrophosphate on Hydroxyapatite and Its Implications in Calcium Homeostasis. Nat. Cell Biol. 1966, 212, 901–903.
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the Mouse ank Gene in Control of Tissue Calcification and Arthritis. Science 2000, 289, 265–270.
- Morava, E.; Kühnisch, J.; Drijvers, J.M.; Robben, J.H.; Cremers, C.; Van Setten, P.; Branten, A.; Stumpp, S.; De Jong, A.; Voesenek, K.; et al. Autosomal Recessive Mental Retardation, Deafness, Ankylosis, and Mild Hypophosphatemia Associated with a Novel ANKH Mutation in a Consanguineous Family. J. Clin. Endocrinol. Metab. 2011, 96, E189–E198.
- Omelon, S.; Georgiou, J.; Henneman, Z.J.; Wise, L.M.; Sukhu, B.; Hunt, T.; Wynnyckyj, C.; Holmyard, D.; Bielecki, R.; Grynpas, M.D. Control of Vertebrate Skeletal Mineralization by Polyphosphates. PLoS ONE 2009, 4, e5634.
- Omelon, S.; Ariganello, M.; Bonucci, E.; Grynpas, M.; Nanci, A. A Review of Phosphate Mineral Nucleation in Biology and Geobiology. Calcif. Tissue Int. 2013, 93, 382–396.
- Jahnen-Dechent, W.; Schäfer, C.; Ketteler, M.; McKee, M.D. Mineral chaperones: A role for fetuin-A and osteopontin in the inhibition and regression of pathologic calcification. J. Mol. Med. 2007, 86, 379–389.
- Jahnen-Dechent, W.; Heiss, A.; Schäfer, C.; Ketteler, M. Fetuin-A Regulation of Calcified Matrix Metabolism. Circ. Res. 2011, 108, 1494–1509.
- Bozycki, L.; Mroczek, J.; Bessueille, L.; Mebarek, S.; Buchet, R.; Pikula, S.; Strzelecka-Kiliszek, A. Annexins A2, A6 and Fetuin-A Affect the Process of Mineralization in Vesicles Derived from Human Osteoblastic hFOB 1.19 and Osteosarcoma Saos-2 Cells. Int. J. Mol. Sci. 2021, 22, 3993.
- Seto, J.; Busse, B.; Gupta, H.S.; Schäfer, C.; Krauss, S.; Dunlop, J.; Masic, A.; Kerschnitzki, M.; Zaslansky, P.; Boesecke, P.; et al. Accelerated Growth Plate Mineralization and Foreshortened Proximal Limb Bones in Fetuin-A Knockout Mice. PLoS ONE 2012, 7, e47338.
- Cai, M.M.X.; Smith, E.R.; Holt, S.G. The role of fetuin-A in mineral trafficking and deposition. BoneKEy Rep. 2015, 4, 672.
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural Basis of Calcification Inhibition by α2-HS Glycoprotein/Fetuin-A. J. Biol. Chem. 2003, 278, 13333–13341.
- George, A.; Veis, A. Phosphorylated Proteins and Control over Apatite Nucleation, Crystal Growth, and Inhibition. Chem. Rev. 2008, 108, 4670–4693.
- Beniash, E.; Deshpande, A.S.; Fang, P.A.; Lieb, N.S.; Zhang, X.; Sfeir, C.S. Possible role of DMP1 in dentin mineralization. J. Struct. Biol. 2011, 174, 100–106.
- Ling, Y.; Rios, H.F.; Myers, E.R.; Lu, Y.; Feng, J.Q.; Boskey, A.L. DMP1 depletion decreases bone mineralization in vivo: An FTIR imaging analysis. J. Bone Miner. Res. 2005, 20, 2169–2177.
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315.
- Gericke, A.; Qin, C.; Spevak, L.; Fujimoto, Y.; Butler, W.T.; Sørensen, E.S.; Boskey, A.L. Importance of Phosphorylation for Osteopontin Regulation of Biomineralization. Calcif. Tissue Int. 2005, 77, 45–54.
- Kaipatur, N.; Murshed, M.; McKee, M. Matrix Gla Protein Inhibition of Tooth Mineralization. J. Dent. Res. 2008, 87, 839–844.
- Holm, E.; Gleberzon, J.S.; Liao, Y.; Sørensen, E.S.; Beier, F.; Hunter, G.K.; Goldberg, H.A. Osteopontin mediates mineralization and not osteogenic cell development In Vitro. Biochem. J. 2014, 464, 355–364.
- Marinovich, R.; Soenjaya, Y.; Wallace, G.Q.; Zuskov, A.; Dunkman, A.; Foster, B.L.; Ao, M.; Bartman, K.; Lam, V.; Rizkalla, A.; et al. The role of bone sialoprotein in the tendon–bone insertion. Matrix Biol. 2016, 52–54, 325–338.
- Boskey, A.; Spevak, L.; Paschalis, E.; Doty, S.; McKee, M. Osteopontin Deficiency Increases Mineral Content and Mineral Crystallinity in Mouse Bone. Calcif. Tissue Int. 2002, 71, 145–154.
- Hruska, K.A. Vascular Smooth Muscle Cells in the Pathogenesis of Vascular Calcification. Circ. Res. 2009, 104, 710–711.
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81.
- Addison, W.N.; Nakano, Y.; Loisel, T.; Crine, P.; McKee, M.D. MEPE-ASARM Peptides Control Extracellular Matrix Mineralization by Binding to Hydroxyapatite: An Inhibition Regulated by PHEX Cleavage of ASARM. J. Bone Miner. Res. 2008, 23, 1638–1649.
- Salmon, B.; Bardet, C.; Khaddam, M.; Naji, J.; Coyac, B.; Baroukh, B.; Letourneur, F.; Lesieur, J.; Decup, F.; Le Denmat, D.; et al. MEPE-Derived ASARM Peptide Inhibits Odontogenic Differentiation of Dental Pulp Stem Cells and Impairs Mineralization in Tooth Models of X-Linked Hypophosphatemia. PLoS ONE 2013, 8, e56749.
- Gowen, L.C.; Petersen, D.N.; Mansolf, A.L.; Qi, H.; Stock, J.L.; Tkalcevic, G.T.; Simmons, H.A.; Crawford, D.T.; Chidsey-Frink, K.L.; Ke, H.Z.; et al. Targeted Disruption of the Osteoblast/Osteocyte Factor 45 Gene (OF45) Results in Increased Bone Formation and Bone Mass. J. Biol. Chem. 2003, 278, 1998–2007.
- Quarles, L.D. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am. J. Physiol. Metab. 2003, 285, E1–E9.
- Orimo, H. Hypophosphatasia: A systemic skeletal disorder caused by alkaline phosphatase deficiency. In Pathophysiology-Altered Physiological States; Gaze, D.C., Ed.; IntechOpen: London, UK, 2018.
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246.
- Meah, F.; Basit, A.; Emanuele, N.; Emanuele, M.A. Hypophosphatasia: Review of Bone Mineral Metabolism, Pathophysiology, Clinical Presentation, Diagnosis, and Treatment. Clin. Rev. Bone Miner. Metab. 2017, 15, 24–36.
- Di Mauro, S.; Manes, T.; Hessle, L.; Kozlenkov, A.; Pizauro, J.M.; Hoylaerts, M.F.; Millán, J.L. Kinetic Characterization of Hypophosphatasia Mutations with Physiological Substrates. J. Bone Miner. Res. 2002, 17, 1383–1391.
- Whyte, M.P. Hypophosphatasia: Enzyme Replacement Therapy Brings New Opportunities and New Challenges. J. Bone Miner. Res. 2017, 32, 667–675.
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-Replacement Therapy in Life-Threatening Hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913.
- Liedtke, D.; Hofmann, C.; Jakob, F.; Klopocki, E.; Graser, S. Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules 2020, 10, 1648.
- Yadav, M.C.; Simão, A.M.S.; Narisawa, S.; Huesa, C.; McKee, M.D.; Farquharson, C.; Millán, J.L. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: A unified model of the mechanisms of initiation of skeletal calcification. J. Bone Miner. Res. 2011, 26, 286–297.
- Huesa, C.; Houston, D.; Kiffer-Moreira, T.; Yadav, M.C.; Millan, J.L.; Farquharson, C. The functional co-operativity of tissue-nonspecific alkaline phosphatase (TNAP) and PHOSPHO1 during initiation of skeletal mineralization. Biochem. Biophys. Rep. 2015, 4, 196–201.
- Terkeltaub, R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2006, 2, 371–377.
- Stover, P.J.; Field, M. Vitamin B-6. Adv. Nutr. 2015, 6, 132–133.
- Sebastián-Serrano, Á.; de Diego-García, L.; Martínez-Frailes, C.; Avila, J.; Zimmermann, H.; Millán, J.L.; Miras-Portugal, M.T.; Díaz-Hernández, M. Tissue-nonspecific Alkaline Phosphatase Regulates Purinergic Transmission in the Central Nervous System During Development and Disease. Comput. Struct. Biotechnol. J. 2015, 13, 95–100.
- Baumgartner-Sigl, S.; Haberlandt, E.; Mumm, S.; Scholl-Bürgi, S.; Sergi, C.; Ryan, L.; Ericson, K.L.; Whyte, M.P.; Högler, W. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone 2007, 40, 1655–1661.
- Coburn, S.P.; Whyte, M.P. Role of phosphatases in the regulation of vitamin B-6 metabolism in hypophosphatasia and other disorders. In Current Topics in Nutrition and Disease Volume 19: Clinical and Physiological Application of Vitamin B-6; Leklem, J.E., Reynolds, R.D., Eds.; Alan R. Liss, Inc.: New York, NY, USA, 1988; pp. 65–93.
- Narisawa, S.; Hasegawa, H.; Watanabe, K.; Millán, J.L. Stage-specific expression of alkaline phosphatase during neural development in the mouse. Dev. Dyn. 1994, 201, 227–235.
- Fonta, C.; Negyessy, L. Subcellular biochemistry. In Neuronal Tissue-Nonspecific Alkaline Phosphatase (TNAP); Springer: Dordrecht, The Netherlands, 2015.
- Narisawa, S.; Fröhlander, N.; Millán, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446.
- Fedde, K.N.; Whyte, M.P. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridox-al-5′-phosphate ectophosphatase: Normal and hypophosphatasia fibroblast study. Am. J. Hum. Genet. 1990, 47, 767–775.
- Schiroli, D.; Ronda, L.; Peracchi, A. Kinetic characterization of the humanO-phosphoethanolamine phospho-lyase reveals unconventional features of this specialized pyridoxal phosphate-dependent lyase. FEBS J. 2014, 282, 183–199.
- Hunter, G.K.; Kyle, C.L.; Goldberg, H.A. Modulation of crystal formation by bone phosphoproteins: Structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem. J. 1994, 300, 723–728.
- Goldberg, H.A.; Warner, K.J.; Li, M.C.; Hunter, G.K. Binding of Bone Sialoprotein, Osteopontin and Synthetic Polypeptides to Hydroxyapatite. Connect. Tissue Res. 2001, 42, 25–37.
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of Osteopontin Is Required for Inhibition of Vascular Smooth Muscle Cell Calcification. J. Biol. Chem. 2000, 275, 20197–20203.
- Harmey, D.; Johnson, K.A.; Zelken, J.; Camacho, N.P.; Hoylaerts, M.F.; Noda, M.; Terkeltaub, R.; Millán, J.L. Elevated Skeletal Osteopontin Levels Contribute to the Hypophosphatasia Phenotype in Akp2−/− Mice. J. Bone Miner. Res. 2006, 21, 1377–1386.
- Garnero, P.; Delmas, P.D. Assessment of the serum levels of bone alkaline phosphatase with a new immunoradiometric assay in patients with metabolic bone disease. J. Clin. Endocrinol. Metab. 1993, 77, 1046–1053.
- Nizet, A.; Cavalier, E.; Stenvinkel, P.; Haarhaus, M.; Magnusson, P. Bone alkaline phosphatase: An important biomarker in chronic kidney disease—Mineral and bone disorder. Clin. Chim. Acta 2020, 501, 198–206.
- Pettengill, M.; Robson, S.; Tresenriter, M.; Millán, J.L.; Usheva, A.; Bingham, T.; Belderbos, M.; Bergelson, I.; Burl, S.; Kampmann, B.; et al. Soluble Ecto-5′-nucleotidase (5′-NT), Alkaline Phosphatase, and Adenosine Deaminase (ADA1) Activities in Neonatal Blood Favor Elevated Extracellular Adenosine. J. Biol. Chem. 2013, 288, 27315–27326.
- Pettengill, M.; Matute, J.D.; Tresenriter, M.; Hibbert, J.; Burgner, D.; Richmond, P.; Millán, J.L.; Ozonoff, A.; Strunk, T.; Currie, A.; et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS ONE 2017, 12, e0175936.
- Schmidt, T.; Rolvien, T.; Linke, C.; Jandl, N.M.; Oheim, R.; Amling, M.; Barvencik, F. Outcome of Teriparatide Treatment on Fracture Healing Complications and Symptomatic Bone Marrow Edema in Four Adult Patients with Hypophosphatasia. JBMR Plus 2019, 3, e10215.
- Whyte, M.P.; Wenkert, D.; McAlister, W.H.; Mughal, M.Z.; Freemont, A.J.; Whitehouse, R.; Baildam, E.M.; Coburn, S.P.; Ryan, L.M.; Mumm, S. Chronic Recurrent Multifocal Osteomyelitis Mimicked in Childhood Hypophosphatasia. J. Bone Miner. Res. 2009, 24, 1493–1505.
- Abate, M.; Salini, V.; Andia, I. Tendons Involvement in Congenital Metabolic Disorders. Adv. Exp. Med. Biol. 2016, 920, 117–122.
- Kapferer-Seebacher, I.; Foradori, L.; Zschocke, J.; Schilke, R. Rare Genetic Disorders Affecting the Periodontal Supporting Tissues in Adolescence. Front. Dent. Med. 2021, 2.
- Bessueille, L.; Briolay, A.; Como, J.; Mebarek, S.; Mansouri, C.; Gleizes, M.; El Jamal, A.; Buchet, R.; Dumontet, C.; Matera, E.; et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 2020, 133, 115262.
- Beck, C.; Morbach, H.; Richl, P.; Stenzel, M.; Girschick, H.J. How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol. Int. 2008, 29, 229–238.
- Mulay, S.R.; Steiger, S.; Shi, C.; Anders, H. A guide to crystal-related and nano- or microparticle-related tissue responses. FEBS J. 2019, 287, 818–832.
- Zhang, X.; Shu, Q.; Liu, Z.; Gao, C.; Wang, Z.; Xing, Z.; Song, J. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. 2021, 1751, 147170.
- Hernández-Chirlaque, C.; Gámez-Belmonte, R.; Ocón, B.; Martínez-Moya, P.; Wirtz, S.; de Medina, F.S.; Martínez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohns Coliti 2016, 11, 857–870.
- Vaughan, K.R.; Stokes, L.; Prince, L.; Marriott, H.; Meis, S.; Kassack, M.U.; Bingle, C.; Sabroe, I.; Surprenant, A.; Whyte, M.K.B. Inhibition of Neutrophil Apoptosis by ATP Is Mediated by the P2Y11Receptor. J. Immunol. 2007, 179, 8544–8553.
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP Release Guides Neutrophil Chemotaxis via P2Y2 and A3 Receptors. Science 2006, 314, 1792–1795.
- Hümmeke-Oppers, F.; Hemelaar, P.; Pickkers, P. Innovative Drugs to Target Renal Inflammation in Sepsis: Alkaline Phosphatase. Front. Pharmacol. 2019, 10.
- Vorbrodt, A.W.; Lossinsky, A.S.; Wisniewski, H.M. Localization of Alkaline Phosphatase Activity in Endothelia of Developing and Mature Mouse Blood-Brain Barrier. Dev. Neurosci. 1986, 8, 1–13.
- Bell, M.A.; Scarrow, W.G. Staining for microvascular alkaline phosphatase in thick celloidin sections of nervous tissue: Morphometric and pathological applications. Microvasc. Res. 1984, 27, 189–203.
- Langer, D.; Ikehara, Y.; Takebayashi, H.; Hawkes, R.; Zimmermann, H. The ectonucleotidases alkaline phosphatase and nucleoside triphosphate diphosphohydrolase 2 are associated with subsets of progenitor cell populations in the mouse embryonic, postnatal and adult neurogenic zones. Neuroscience 2007, 150, 863–879.
- Deracinois, B.; Lenfant, A.-M.; Dehouck, M.-P.; Flahaut, C. Tissue Non-specific Alkaline Phosphatase (TNAP) in Vessels of the Brain. Subcell. Biochem. 2015, 76, 125–151.
- Fonta, C.; Barone, P.; Martinez, L.R.; Négyessy, L. Rediscovering TNAP in the Brain: A Major Role in Regulating the Function and Development of the Cerebral Cortex. Subcell. Biochem. 2015, 76, 85–106.
- Demirbilek, H.; Alanay, Y.; Alikaşifoğlu, A.; Topçu, M.; Mornet, E.; Özön, A.; Kandemir, N. Hypophosphatasia Presenting with Pyridoxine-Responsive Seizures, Hypercalcemia, and Pseudotumor Cerebri: Case Report. J. Clin. Res. Pediatric Endocrinol. 2012, 4, 34–38.
- Fukazawa, M.; Tezuka, J.; Sasazuki, M.; Masumoto, N.; Baba, H.; Doi, T.; Tsutsumi, Y.; Mizuno, Y.; Mihara, F.; Nakayama, H. Infantile hypophosphatasia combined with vitamin B6-responsive seizures and reticular formation lesions on magnetic resonance imaging: A case report. Brain Dev. 2018, 40, 140–144.
- Ermonval, M.; Baudry, A.; Baychelier, F.; Pradines, E.; Pietri, M.; Oda, K.; Schneider, B.; Mouillet-Richard, S.; Launay, J.-M.; Kellermann, O. The Cellular Prion Protein Interacts with the Tissue Non-Specific Alkaline Phosphatase in Membrane Microdomains of Bioaminergic Neuronal Cells. PLoS ONE 2009, 4, e6497.
- Vardy, E.R.; Kellett, K.; Cocklin, S.L.; Hooper, N. Alkaline Phosphatase Is Increased in both Brain and Plasma in Alzheimer’s Disease. Neurodegener. Dis. 2012, 9, 31–37.
- Diaz-Hernandez, M.; Gómez-Ramos, A.; Rubio, A.; Gomez-Villafuertes, R.; Naranjo, J.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific Alkaline Phosphatase Promotes the Neurotoxicity Effect of Extracellular Tau. J. Biol. Chem. 2010, 285, 32539–32548.
- Brichacek, A.L.; Benkovic, S.A.; Chakraborty, S.; Nwafor, D.C.; Wang, W.; Jun, S.; Dakhlallah, D.; Geldenhuys, W.J.; Pinkerton, A.B.; Millán, J.L.; et al. Systemic inhibition of tissue-nonspecific alkaline phosphatase alters the brain-immune axis in experimental sepsis. Sci. Rep. 2019, 9, 18788.
- Petty, G.W.; Brown, R.D., Jr.; Whisnant, J.P.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Ischemic Stroke Subtypes. Stroke 1999, 30, 2513–2516.
- Ryu, W.-S.; Lee, S.-H.; Kim, C.K.; Kim, B.J.; Yoon, B.-W. Increased serum alkaline phosphatase as a predictor of long-term mortality after stroke. Neurology 2010, 75, 1995–2002.
- Uehara, T.; Ohara, T.; Minematsu, K.; Nagatsuka, K.; Toyoda, K. Predictors of Stroke Events in Patients with Transient Ischemic Attack Attributable to Intracranial Stenotic Lesions. Intern. Med. 2018, 57, 295–300.
- Zhong, C.; You, S.; Chen, J.; Zhai, G.; Du, H.; Luo, Y.; Dong, X.; Cao, Y.; Liu, C.-F.; Zhang, Y. Serum Alkaline Phosphatase, Phosphate, and In-Hospital Mortality in Acute Ischemic Stroke Patients. J. Stroke Cerebrovasc. Dis. 2018, 27, 257–266.
- Liu, Y.; Liang, X.; Xu, X.; Dong, M.; Jia, S.; Lu, C.; Wei, Y. Increased Serum Alkaline Phosphatase in Patients with Acute Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2018, 28, 21–25.
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317.
- Hoshi, K.; Amizuka, N.; Oda, K.; Ikehara, Y.; Ozawa, H. Immunolocalization of tissue non-specific alkaline phosphatase in mice. Histochem. Cell Biol. 1997, 107, 183–191.
- Suzuki, N.; Irie, M.; Iwata, K.; Nakane, H.; Yoshikane, M.; Koyama, Y.; Uehara, Y.; Takeyama, Y.; Kitamura, Y.; Sohda, T. Altered expression of alkaline phosphatase (ALP) in the liver of primary biliary cirrhosis (PBC) patients. Hepatol. Res. 2006, 35, 37–44.
- Alvaro, D.; Benedetti, A.; Marucci, L.; Monache, M.D.; Monterubbianesi, R.; Di Cosimo, E.; Perego, L.; Macarri, G.; Glaser, S.; Le Sage, G.; et al. The function of alkaline phosphatase in the liver: Regulation of intrahepatic biliary epithelium secretory activities in the rat. Hepatology 2000, 32, 174–184.
- Mimura, Y.; Sakisaka, S.; Harada, M.; Sata, M.; Tanikawa, K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology 1995, 109, 1969–1976.
- Nouwen, E.J.; De Broe, M.E. Human intestinal versus tissue-nonspecific alkaline phosphatase as complementary urinary markers for the proximal tubule. Kidney Int. Suppl. 1994, 47, S43–S51.
- Moochhala, S.H.; Sayer, J.A.; Carr, G.; Simmons, N.L. Renal calcium stones: Insights from the control of bone mineralization. Exp. Physiol. 2008, 93, 43–49.
- Nitschke, Y.; Rutsch, F. Inherited Arterial Calcification Syndromes: Etiologies and Treatment Concepts. Curr. Osteoporos. Rep. 2017, 15, 255–270.
- Opdebeeck, B.; Neven, E.; Millán, J.; Pinkerton, A.; D’Haese, P.; Verhulst, A. Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme. Pharmaceutics 2021, 13, 1138.
- Jaminon, A.M.G.; Dai, L.; Qureshi, A.R.; Evenepoel, P.; Ripsweden, J.; Söderberg, M.; Witasp, A.; Olauson, H.; Schurgers, L.J.; Stenvinkel, P. Matrix Gla protein is an independent predictor of both intimal and medial vascular calcification in chronic kidney disease. Sci. Rep. 2020, 10, 6586.
- Doherty, T.M.; Fitzpatrick, L.A.; Inoue, D.; Qiao, J.-H.; Fishbein, M.C.; Detrano, R.C.; Shah, P.K.; Rajavashisth, T.B. Molecular, Endocrine, and Genetic Mechanisms of Arterial Calcification. Endocr. Rev. 2004, 25, 629–672.
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817.
- Greenland, P. Coronary Artery Calcium Score Combined with Framingham Score for Risk Prediction in Asymptomatic Individuals. Jama 2004, 291, 210–215.
- Patel, J.J.; Bourne, L.E.; Davies, B.K.; Arnett, T.R.; MacRae, V.E.; Wheeler-Jones, C.P.; Orriss, I.R. Differing calcification processes in cultured vascular smooth muscle cells and osteoblasts. Exp. Cell Res. 2019, 380, 100–113.
- Alves, R.D.; Eijken, M.; van de Peppel, J.; van Leeuwen, J.P. Calcifying vascular smooth muscle cells and osteoblasts: Independent cell types exhibiting extracellular matrix and biomineralization-related mimicries. BMC Genom. 2014, 15, 1–14.
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular Calcification. Circ. Res. 2006, 99, 1044–1059.
- Roszkowska, M. Molecular Mechanisms of Vascular Smooth Muscle Cell Trans-Differentiation and Calcification in Atherosclerosis. Ph.D. Thesis, Université de Lyon, Lyon, France, 2018.
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4.
- Romanelli, F.; Corbo, A.; Salehi, M.; Yadav, M.C.; Salman, S.; Petrosian, D.; Rashidbaigi, O.J.; Chait, J.; Kuruvilla, J.; Plummer, M.; et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS ONE 2017, 12, e0186426.
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced Plasma Pyrophosphate Levels in Hemodialysis Patients. J. Am. Soc. Nephrol. 2005, 16, 2495–2500.
- Laurain, A.; Rubera, I.; Duranton, C.; Rutsch, F.; Nitschke, Y.; Ray, E.; Vido, S.; Sicard, A.; Lefthériotis, G.; Favre, G. Alkaline Phosphatases Account for Low Plasma Levels of Inorganic Pyrophosphate in Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8.
- Villa-Bellosta, R. New insights into endogenous mechanisms of protection against arterial calcification. Atherosclerosis 2020, 306, 68–74.
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.-C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517.
- Ziegler, S.G.; Gahl, W.A.; Ferreira, C.R. Generalized Arterial Calcification of Infancy; University of Washington: Seattle, WA, USA, 2020.
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ’idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381.
- Neven, E.G.; De Broe, M.E.; D’Haese, P.C. Prevention of vascular calcification with bisphosphonates without affecting bone mineralization: A new challenge? Kidney Int. 2009, 75, 580–582.
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikuła, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 643–653.
- Villa-Bellosta, R.; Wang, X.; Millán, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Circ. Physiol. 2011, 301, H61–H68.
- Narisawa, S.; Harmey, D.; Yadav, M.C.; O’Neill, W.C.; Hoylaerts, M.F.; Millán, J.L. Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification. J. Bone Miner. Res. 2007, 22, 1700–1710.
- Kiffer-Moreira, T.; Yadav, M.C.; Zhu, N.; Narisawa, S.; Sheen, C.; Stec, B.; Cosford, N.D.; Dahl, R.; Farquharson, C.; Hoylaerts, M.F.; et al. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J. Bone Miner. Res. 2012, 28, 81–91.
More