Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Bruce Ren and Version 1 by Svetlana Sharifulina.
Cerebral ischemia is the second leading cause of death in the world and multimodal stroke therapy is needed. The ischemic stroke generally reduces the gene expression due to suppression of acetylation of histones H3 and H4. Histone deacetylases inhibitors have been shown to be effective in protecting the brain from ischemic damage. Histone deacetylases inhibitors induce neurogenesis and angiogenesis in damaged brain areas promoting functional recovery after cerebral ischemia.
- ischemic stroke
- epigenetics
- histone deacetylase
- histone deacetylase inhibitor
1. Ischemic Stroke Treatment Challenges
Stroke is one of the leading causes of death in the world [1]. About 5 million people die every year. No more than 20% of surviving patients can return to their previous job. 2/3 of strokes occur in people over 65. However, strokes are getting younger. In recent years, about 20% of cerebrovascular accidents have been reported in people aged fewer than 50, and this number is steadily increasing. Thus, the stroke problem is of extreme medical and social importance [2,3,4,5,6,7][2][3][4][5][6][7].
In ischemic stroke (about 80% of all strokes) the occlusion of cerebral arteries by a thrombus, atherosclerotic plaque, spasm, or abrupt changes in blood pressure sharply decreases or stops the blood supply of the brain tissue [4,5,6,8][4][5][6][8]. Cerebral ischemia may be the presenting manifestation of hematological diseases and hematological disorders account for about 1.3% of all causes of acute stroke [9,10][9][10].
Stroke is a developing over time multi-stage process. It starts from the primary minor changes and leads to the formation of a penumbra, death or restoration of its cells, and to the irreversible structural damage of brain tissue causing neurodegeneration. A huge number of pathophysiological and biochemical processes in intracellular and intercellular signaling, proteolysis, and regulation of the transcriptional activity of the genome are occurred in between these phases. Each of these stages is the time point of possible application of anti-stroke drugs. Many compounds tested in animal and cell models with the exception of tissue plasminogen activator (tPA) or endovascular thrombectomy [11] reduced apoptotic cell counts, increased infarction size, and improved neurological deficits after stroke [12,13,14][12][13][14]. However, none of these drugs have been successful in clinical trials. Probably, the complex pathophysiology of cerebral ischemia, the complexity of signaling cascades and differences in the mechanisms of the acute and restorative phases of stroke are the main reasons for failures in translating experimental studies into clinical practice.
Numerous studies searching targets for neuroprotection have highlighted the importance of multimodal stroke therapy. An example of such a strategy, which has been shown to be effective in various models of ischemia, is the inhibition of histone deacetylases (HDACs) [15,16,17,18,19][15][16][17][18][19]. This review analyzes data on the neuroprotective activity of nonspecific HDACs inhibitors (iHDACs) and selective iHDACs.
2. Histone Deacetylases
Cerebral ischemia generally reduces the global level of gene expression due to suppression of acetylation of histones H3 and H4 [20,21][20][21]. The antibody microarray study and immunofluorescence microscopy have shown the twofold decrease in the acetylation of lysine 9 in histone H3 (H3K9Ac) in the ischemic penumbra at 1 h after photothrombotic stroke in the rat brain cortex, and more than fourfold decrease at 4 and 24 h. H3K9Ac was shown to localize exclusively in the neuronal, but not astroglial nuclei. These effects could be associated either with downregulation of histone acetyltransferases, or with overexpression of histone deacetylases [22]. Histone acetyltransferases (HATs) acetylate lysine residues in the histone tails. This promotes DNA unfolding and chromatin decondensation that facilitates transcription and protein synthesis. Histone deacetylases remove the acetyl groups from histones. This leads to formation of the transcriptionally inactive heterochromatin, in which gene expression is hindered. To maintain the transcriptionally active state of chromatin, HATs and HDACs should work together [23,24,25][23][24][25].
Among a large number of cellular non-histone proteins deacetylated by HDACs are transcription factors and co-regulators (e.g., c-MYC, HMG, YY1, EKLF, E2F1, factors GATA, HIF-1α, MyoD, NF-κB, and FoxB3), tumor suppressor proteins (e.g., p53, RUNX3), signaling mediators (e.g., STAT 1 and 3, β-catenin, and SMAD7), steroid receptors (e.g., androgens, estrogen, and SHP), and chaperone proteins and nuclear transport proteins (e.g., α-tubulin, importin-α, cortactin, Ku70, and HSP90) [26,27,28][26][27][28]. These proteins determine the growth, differentiation, migration, and activity of the protein determining cell survival both in normal conditions and under damage [27]. Thus, deacetylation-dependent signaling pathways play a crucial role in cell homeostasis.
Four classes of HDACs are distinguished in mammals according to their functions, intracellular localization, and expression patterns (Figure 1). Class I includes HDAC1, HDAC2, HDAC3, and HDAC8, class II consists of HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10), and class IV contains only one HDAC11. All of them are zinc-dependent enzymes. Sirtuins use nicotinamide adenine dinucleotide NAD+ as a cofactor. They form the class III histone deacetylases. HDACs are evolutionary conservative [29,30,31,32][29][30][31][32].
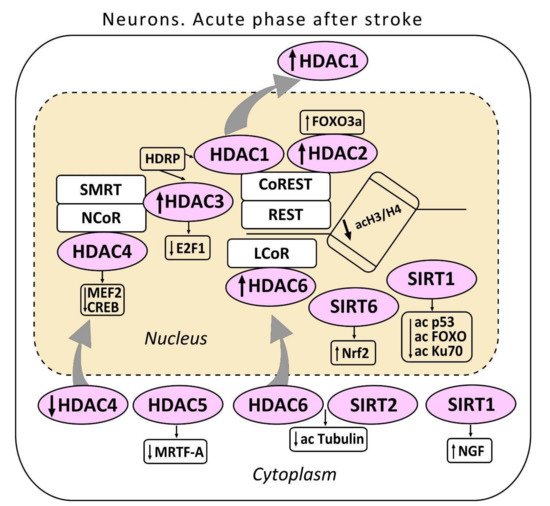
Figure 1. The acute phase after stroke in the neuron. The localization of histone deacetylases (HDAC 1-6) including sirtuins (SIRT 1,2) in the cell is shown. HDAC3 and HDAC1 interact with histone deacetylase-related protein (HDRP), a shortened form of HDAC9. HDRP inhibited the HDAC1/HDAC3 interaction. HDAC1 and HDAC2 are included in the repressor element-1 silencing transcription factor (REST) corepressor 1 (CoREST) complex. HDAC3 in the nuclear receptor co-repressor (NCOR)/silencing mediator for retinoid or thyroid-hormone receptors (SMRT) complex suppresses the production of the pro-apoptotic transcription factor E2F1. HDAC4 suppresses the activity of myocyte enhancer factor 2 (MEF2) and cAMP response element-binding protein (CREB). HDAC4 interacts with the co-repressor complex HDAC3/NCOR. HDAC5 suppressed myocardial transcription factor-A (MRTF-A). One of the cytoplasmic substrates of HDAC6 - α-tubulin is shown. SIRT1 deacetylates p53, FOXO, and Ku70 proteins. The cytoplasmic SIRT1 induces nerve growth factor NGF. SIRT6 stimulates the nuclear factor erythroid 2–related factor (Nrf2).
3. Role of Histone Deacetylases in Cell Damage and Recovery after Cerebral Ischemia
3.1. Class I HDACs
The first class HDACs is widely represented in the brain [31]. HDAC1 localizes both in the neuronal nuclei, and in the cytoplasm, where it deacetylates some cytoplasmic proteins. HDAC2 localizes exclusively in the neuronal nuclei [22,33][22][33]. HDAC1 suppresses the production of proteins, which regulate the cell cycle in somatic cells. It also contributes to cell protection against DNA damage [34]. HDAC1 can serve as a molecular switch between neuronal survival and death [35]. HDAC2 regulates apoptosis in the ischemic penumbra [33,36][33][36]. HDAC1 and HDAC2 can be included in the multienzyme complexes Sin3, NuRD, CoREST, or NODE that suppress transcription of different sets of target genes [37,38,39][37][38][39]. The complex CoREST suppresses genes involved in synaptic plasticity and post-stroke recovery [40,41][40][41] (Figure 1).
The upregulation of HDAC2, but not HDAC1, in the PTS-induced penumbra was associated with development of apoptosis [22]. Other authors also showed the critical role of HDAC2 in death of neurons in the peri-infarction area after ischemic stroke. The upregulation of HDAC2 observed in the early recovery phase from five to seven post-stroke days reduced survival of neurons and augment neuroinflammation. HDAC2 targeting is apparently a novel therapeutic strategy for stroke recovery [36]. MCAO-induced overexpression of HDAC2 decreased the number of synapses, impaired synaptic plasticity, reduced memory, and deteriorated other cerebral functions [42,43][42][43]. Knockdown or knockout of the HDAC2 gene restored brain functions due to plasticity of the surviving neurons in the peri-infarction zone [43,44][43][44].
The overexpression of HDAC1 and HDAC2 in ischemic penumbra neurons and white matter glial cells was observed in the mouse brain during the early regeneration period, 1 week after MCAO. Their levels in the infarct core, oppositely, decreased [31]. Long-term overexpression of HDAC2 and HDAC8 was observed in neurons and astrocytes at 3‒14 days after photothrombotic stroke in the mouse cerebral cortex [45]. Thus, HDAC2 deteriorates synaptic processes, impairs memory, disturbs various cerebral functions, and stimulates apoptosis in the ischemic brain.
HDAC3 also deacetylates histones H3 and H4 and some non-histone proteins. It also contributes to regulation of DNA replication and repair. The complex HDAC3/NCOR/SMRT is essential for maintaining chromatin structure and genome stability [46]. Overexpression of HDAC3, HDAC6, and HDAC11 was observed in the ischemic penumbra 3 and 24 h after MCAO, and persisted for a week after reperfusion. The upregulation of HDAC3 and HDAC6 in the mouse cortical neurons was also observed in vitro in the neuronal cell culture. The inhibition of HDAC3 or HDAC6 expression by the short hairpin shRNA increased cell viability. This suggested their involvement in ischemia-induced neurotoxicity [47]. The ischemia-induced neurotoxicity of HDAC3 was demonstrated in other studies [35,48,49][35][48][49]. The neurotoxic effect of HDAC3 was associated with its binding to HDAC1. Actually, the knockdown of HDAC3 suppressed the neurotoxicity of HDAC1, whereas HDAC1 knockdown suppressed the neurotoxicity of HDAC3. HDAC3 and HDAC1 interact with histone deacetylase-related protein (HDRP), a shortened form of HDAC9, whose expression was reduced during neuronal death. The interaction between HDRP and HDAC1, but not HDAC3 protected neurons. HDRP inhibited the HDAC1/HDAC3 interaction and prevented the neurotoxic effect of any of these proteins. This is a possible mechanism of HDAC1-mediated action as a switch “survival/death” in cerebral neurons. HDAC1 interaction with HDRP promotes neuron survival, whereas its interaction with HDAC3 leads to apoptosis [50]. On the other hand, HDAC3 was shown to suppress the production of the pro-apoptotic transcription factor E2F1 in neurons, and thus to contribute to survival of these cells [51].
Another class I histone deacetylase HDAC8 is present mainly in the cytoplasm of neurons and astrocytes of the cerebral cortex, amygdala, hippocampus, and hypothalamus [45,52][45][52]. The expression of HDAC8 in the mouse cortical neurons and astrocytes increased significantly during the recovery period, from 3 to 14 days after photothrombotic stroke [43].
3.2. Class II HDACs
The data on the role of HDAC4 in neurodegeneration and neuroprotection are contradictory. On one hand, some authors have reported the ability of HDAC4 to maintain neuronal survival [53,54,55][53][54][55]. However, other authors did not find a dependence of neuronal survival on HDAC4 expression [56,57][56][57]. In cultured neurons, HDAC4 rapidly translocates into the nucleus under glutamate release, or decreased K+ concentration in the medium. This stimulated cell death [58,59][58][59]. The administration of brain-derived neurotrophic factor (BDNF) prevented nuclear translocation of HDAC4 [60]. On the contrary, inhibition or loss of calmodulin-dependent kinase IV (CaMKIV) stimulated HDAC4 accumulation in the neuronal nuclei [59,61][59][61]. Nuclear HDAC4 was shown to promote neuronal apoptosis by suppressing the activity of prosurvival transcription factors MEF2 (myocyte enhancer factor 2) and CREB (cAMP response element-binding protein) [58]. Other authors have reported that HDAC4 translocation into the nuclei of neurons, but not astrocytes, did not cause apoptosis in the MCAO-induced ischemic penumbra. Moreover, the nuclear localization of HDAC4 promoted post-stroke brain recovery [62]. The HDAC4 level in the cytoplasmic, but not nuclear fraction of the rat brain cortex decreased at 24 h after photothrombotic stroke [33]. The downregulation of HDAC4 and its relocalization into the neuronal nuclei continued during the recovery period, 2 weeks after stroke [63,64][63][64]. In the neuronal nuclei HDAC4 deacetylates histones H3 and H4 and decreases the levels of some prosurvival proteins that finally lead to the neuronal death [58,59,64][58][59][64]. Since HDAC4 was assumed to be inactive against histones, these effects could be mediated by its interaction with other nuclear proteins. Actually, HDAC4 was shown to exhibit deacetylase activity after interacting with the co-repressor complex HDAC3/NCOR [65]. Further studies of HDAC4 interactions with different proteins are needed to understand its role in survival and death of cerebral cells after stroke.
HDAC5, another member of class II histone deacetylases, is involved in neuronal differentiation and axon regeneration in the injured sensory neurons [66,67][66][67]. The overexpression of HDAC5 and its nuclear localization was shown to be associated with apoptosis of the cultured neurons from the cerebellar granular layer [68]. After transient MCAO, HDAC5 suppressed the antiapoptotic effect of the transcription factor MRTF-A (myocardial transcription factor-A) in the rat brain neurons [69]. HDAC5 expression in the ischemic penumbra decreased 1, 2, and 14 days after MCAO [47,64][47][64]. The downregulation of HDAC5 in the mouse cerebral cortex was observed at 3 days after photothrombotic stroke. However, the number of the apoptotic HDAC5-positive cells did not change [63]. Possibly, the decrease in the level of HDAC5 in cortical neurons was associated with the regeneration processes [70].
HDAC6 belongs to the IIb class of histone deacetylases. It is involved in various cellular processes such as degradation of damaged proteins, cell migration, and intercellular interactions [71]. One of the cytoplasmic substrates of HDAC6 is α-tubulin. Its deacetylation induced destabilization of microtubules in the course of cytoskeleton reorganization and axonal growth during post-stroke regeneration [72]. In the mouse or rat brains HDAC6 presents not only in the cytoplasm, but also in the nuclei of some cortical neurons, but not astrocytes [33,63][33][63]. During the first two weeks after the photothrombotic stroke, HDAC6 was upregulated in the neurons not only in the penumbra, but also in the contralateral cerebral cortex, where it appeared in the neuronal nuclei. In the PTS-induced penumbra, HDAC6 co-localized with apoptotic neurons that indicated its involvement in neuronal apoptosis [57,63][57][63].
Thus, HDACs are widely represented in the brain. The expression of HDAC1, HDAC2, HDAC3, HDAC4, and HDAC6 increased in the ischemic penumbra. Some of them are located in the neuronal nuclei, some in the cytoplasm, and others—both in the nucleus and cytoplasm. Their functions after ischemic stroke differed. Some HDACs mediate prosurvival processes, whereas others are involved in neurotoxicity. HDAC2 and HDAC6 were apparently involved in apoptosis in the post-ischemic brain.
3.3. Sirtuins
Sirtuins (SIRT) are class III histone deacetylases. The coenzyme nicotinamide adenine dinucleotide (NAD+) makes sirtuins sensitive to metabolic and redox changes [73]. In mammals, seven sirtuins have been identified. Of these, SIRT1 and SIRT6 are localized mainly in the cell nuclei, SIRT7 in the nucleoli, SIRT2 in the cytoplasm, and SIRT3, SIRT4, and SIRT5 are mitochondrial proteins [74]. Sirtuins deacetylate a variety of substrates such as transcription factors, enzymes, and histones. They control diverse biological processes including metabolism, cell growth, aging apoptosis, and autophagy [75]. In the present review we focus on the role of non-mitochondrial SIRT1, SIRT2, and SIRT6 in the brain damage and recovery after ischemic stroke.
SIRT1 content in the brain is higher than in other organs [74]. In the hippocampus it regulates synaptic plasticity and memory. Since SIRT1 deacetylates histones and various transcription factors [72[72][76],76], and, also, has the chaperone-like activity [77], its subcellular localization is of significant importance for its functioning. The presence of the nuclear localization signal (NLS) and the nuclear export signal (NES) in the SIRT1 molecule allows it to shuttle from the nucleus to the cytoplasm and back that was assumed to be required for synaptic plasticity and memory formation [78,79][78][79]. The subcellular location of SIRT1 changed during brain development and in response to physiological and pathological stimuli [79,80][79][80].
SIRT1 mediates neuroprotection after ischemic stroke, traumatic brain injury, and neurodegenerative diseases. It regulates neurogenesis, neurite outgrowth, and gliogenesis, which are involved in postischemic brain regeneration [76,81][76][81]. In the SIRT1 knockout mice, MCAO induced greater cerebral infarction than in control animals [82]. On the contrary, mice overexpressing SIRT1 were more resistant to ischemia than control animals [83]. The activation of SIRT1 by resveratrol reduced the MCAO-induced infarction volume [84]. SIRT1 was overexpressed in the ischemic penumbra 7 days after MCAO in the mouse cerebral cortex [82]. Nuclear SIRT1 was reported to prevent apoptosis by deacetylation of proteins p53 [85], FOXO [86], and Ku70 [87]. On the contrary, SIRT1 localized in the cytoplasm enhanced caspase-dependent cell apoptosis [88]. Nevertheless, the translocation of SIRT1 into the cytoplasm was not associated with cell apoptosis in the peri-infarct area at 7 days after photothrombotic stroke in the mouse cerebral cortex. In this case, the cytoplasmic localization of SIRT1 was associated with the upregulation of synaptophysin and GAP-43 that mediate the axon outgrowth and restoration of synaptic connections [89]. The cytoplasmic SIRT1 was shown to enhance the neurite outgrowth that was induced by nerve growth factor NGF. Oppositely, inhibitors of SIRT1 or SIRT1-siRNA significantly reduced this effect [90].
SIRT2 is expressed predominantly in oligodendrocytes and in the myelin-rich regions of the ischemic brain. It was not found in astrocytes, microglia, or neurons [91]. However, other authors reported the presence of SIRT2 in the cytoplasm of neurons, but not astrocytes in the mouse cerebral cortex [89,92][89][92]. In the cytoplasm, SIRT2 such as HDAC6 regulates the microtubule dynamics through deacetylation of α-tubulin [93]. SIRT2 and HDAC6 can deacetylate α-tubulin either together [94], or separately [93]. Interestingly, the inhibition of SIRT2 increased acetylation of the microtubular α-tubulin mainly in the perinuclear zone, whereas inhibition of HDAC6 caused the general hyperacetylation of microtubules throughout the cell [95]. Although some studies pointed to the pathological role of SIRT2 after cerebral ischemia [76[76][96],96], the functions of SIRT2 in the ischemic brain are possibly more complicated than only pathological or only neuroprotective. Indeed, transient MCAO reduced the expression of SIRT2 and its translocation into the neuronal nuclei that played a neuroprotective role [96]. On the contrary, the overexpression of SIRT2 in the cytoplasm of the cerebellar neurons or in vitro in the differentiated PC12 cell line was shown to induce apoptosis [77,97][77][97]. Sirt2 was shown to mediate the myelin-dependent neuronal dysfunction during the early phase after MCAO in the mouse brain. Notably, the dynamics of Sirt2 mRNA and the protein level after ischemia differed [91].
SIRT6 was found in both: cerebral neurons and astrocytes [89,98][89][98]. It deacetylates mainly the lysine residues 9 and 56 in histone H3 that possibly represses genes associated with aging [99]. The role of SIRT6 in ischemia is still unclear and controversial. On one hand, SIRT6 protected the brain from postischemic reperfusion injury due to stimulation of transcription factor Nrf2 (nuclear factor-like (erythroid 2)-like 2), which regulates the expression of antioxidant proteins and suppresses oxidative stress [100,101][100][101]. SIRT6 was co-expressed with GAP-43, a marker of axon growth and synapse formation, at 14 days after photothrombotic stroke in the mouse cerebral cortex [89]. Whether SIRT6 functions as a part of the multiprotein complex in the postsynaptic membranes [102], or it regulates the neurite growth during the post-stroke recovery phase should be further studied. SIRT6 immunofluorescence was not observed in apoptotic cells in the PTS-induced penumbra [89]. On the other hand, its overexpression in cultured neurons under oxygen and glucose deprivation was associated with necrosis of cortical cells [103].
Thus, sirtuins, SIRT1 and SIRT6, are involved in the postischemic brain regeneration. SIRT1 regulates synaptic plasticity, memory, neuritogenesis, neurogenesis, and gliogenesis. SIRT6 protects neurons and astrocytes from the post ischemic reperfusion injury via stimulation of the transcription factor that regulates the production of antioxidant proteins Nrf2.
The HDAC inhibitors that have been developed to date are capable of inhibiting almost all HDAC isoforms of these four classes with varying degrees of specificity.
4. Pan-Inhibitors of Histone Deacetylases in Cerebral Ischemia
Inhibitors of different histone deacetylases that efficiently protect the brain from ischemic injury belong to two chemical groups: (a) Small carboxylates: valproic acid (VPA), sodium butyrate (SB), and sodium 4-phenylbutyrate (4-PBA), and (b) Hydroxamate-containing compounds: suberoylanilide hydroxamic acid (SAHA, Vorinostat), trichostatin A (TSA), and others. They protect the brain against excitotoxicity, oxidative stress, ER stress, apoptosis, inflammation, and BBB breakdown. They also induce angiogenesis, neurogenesis, and migration of stem cells to the damaged brain regions that improves functional recovery after cerebral ischemia. HDAC inhibitors are the promising neuroprotectors for treating ischemic stroke [15,16,17,18][15][16][17][18].
VPA, a pan-HDAC inhibitor, was shown to reduce brain injury in various stroke models. It improved the functional outcome, and demonstrated the anti-inflammatory activity [104,105,106][104][105][106]. VPA induces different proteins such as NeuroD, Math1, Ngn1, and p15, which contribute to differentiation of neural precursors in the hippocampus [107]. VPA administration during 7 days after MCAO considerably improved the neurological outcome in rats. This effect was associated with enhanced white matter repair and neurogenesis [108,109][108][109]. The VPA treatment increased survival of oligodendrocytes and caused the generation of new oligodendrocytes. These effects were associated with the increased density of myelinated axons in the ischemic boundary around the infarction core. At the molecular level, VPA increased the acetylation of histone H4 and caused overexpression of glutamate transporter 1 (GLT1) in neuroblasts. It also increased the number of new neurons [109]. Prolonged application of VPA during two weeks after MCAO increased the acetylation of histones H3 and H4 in the rat brain, and caused the upregulation of transcription factor HIF-1α (hypoxia-inducible factor-1α) and its downstream pro-angiogenic molecules such as vascular endothelial growth factor (VEGF) and matrix metalloproteinases MMP2 and MMP9. This enhanced the microvessel density and promoted functional recovery [106]. VPA suppressed the nuclear translocation of the NF-κB subunit p65, reduced activity of matrix metalloproteinase MMP9, and restored the BBB integrity that was broken after stroke [110].
Other pan-HDAC inhibitors SB and 4-PBA showed similar neuroprotective activity [111,112,113][111][112][113]. SB stimulated proliferation of neuronal progenitor cells and neurogenesis in the SVZ and DG zones of the rat brain after MCAO. It increased the levels of acetylated histone H3, neural cell adhesion molecule nestin, glial fibrillary acidic protein, transcription factor CREB (phospho-cAMP response element-binding protein), and brain-derived neurotrophic factor (BDNF) that were reduced after cerebral ischemia [111]. The neuroprotective effect of SB was also associated with inhibition of oxidative stress, reduction of BBB permeability, and anti-inflammatory action [110]. Notably, VPA and SB stimulated neurogenesis in the peri-infarct regions during the post-stroke recovery period [108,109,111][108][109][111]. SB effect on proliferation, differentiation, and migration of the neural precursor cells was mediated by the BDNF-TrkB signaling pathway [114]. Microglia-mediated neuroinflammation is an important component of the stroke-induced brain pathology. As shown in vitro, SB reduced the acetylation of histone H3 that was increased in the activated microglial cells. It also altered the transcription of pro-inflammatory genes Tnf-α, Nos2, Stat1, and Il6. Simultaneously, SB induced the expression of anti-inflammatory genes regulated by the IL10/STAT3 pathway. In the microglia of mice subjected to MCAO, SB reduced the expression of pro-inflammatory proteins TNF-α and NOS2, and stimulated expression of the anti-inflammatory mediator IL10. Therefore, HDAC inhibition by SB turned the microglial activity in the ischemic brain from the pro-inflammatory to the anti-inflammatory mechanism [115].
The application of SAHA protected the rat brain from MCAO-induced ischemia [116]. It prevented the ischemia-induced decrease of the histone H3 acetylation level and reduced the infarct volume. It also increased the levels of neuroprotective proteins Hsp70 and Bcl-2 in both control and ischemic brains [117]. The intraperitoneal administration of SAHA at 12 h after transient MCAO reduced the infarction volume in the mouse brain and improved the post-stroke outcome. It reduced the level of pro-inflammatory cytokines and inhibited the microglia-mediated inflammatory response [118].
TSA increased survival of cultured neuronal cells after oxygen and glucose deprivation. It also decreased the stroke-induced infarct volume in the mice brain. These effects were associated with the activation of antioxidant processes. TSA mediated HDAC inhibition and activated the transcription factor Nrf2, which regulates expression of diverse antioxidant proteins. As a result, heme oxygenase 1, NAD(P)H:quinone oxidoreductase 1, and glutamate-cysteine ligase were overexpressed and mediated neuroprotection in the neuronal culture and in the ischemic brain [106,116][106][116].
These HDAC inhibitors are non-selective. The neuroprotective effects of non-selective inhibitors such as TSA, SB, and SAHA have been well reviewed [119,120,121,122][119][120][121][122]. They inhibit a group of HDACs that belong to class I, or II, or both. Some HDACs in these groups are involved in ischemia-induced cell death, whereas others participate in the neuroprotective processes. It is of interest to use more selective inhibitors in order to affect only the pathogenic HDACs in the ischemic brain.
References
- Meng, H.; Jin, W.; Yu, L.; Xu, S.; Wan, H.; He, Y. Protective effects of polysaccharides on cerebral ischemia: A mini-review of the mechanisms. Int. J. Biol. Macromol. 2021, 169, 463–472.
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study. Lancet 2014, 383, 245–255.
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study. Lancet 2016, 388, 1459–1544.
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654.
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. 2018 Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke 2018, 49, e46–e99.
- Powers, W.J. Acute ischemic stroke. N. Engl. J. Med. 2020, 383, 252–260.
- Singh, T.P.; Murphy, S.P.; Weinstein, J.R. Stroke: Basic and clinical. Adv. Neurobiol. 2017, 15, 281–293.
- Pimentel, B.C.; Willeit, J.; Töll, T.; Kiechl, S.; e Melo, T.P.; Canhão, P.; Fonseca, C.; Ferro, J. Etiologic evaluation of ischemic stroke in young adults: A comparative study between two european centers. J. Stroke Cerebrovasc. Dis. 2019, 28, 1261–1266.
- Arboix, A.; Besses, C. Cerebrovascular disease as the initial clinical presentation of haematological disorders. Eur. Neurol. 1997, 37, 207–211.
- Arboix, A.; Jiménez, C.; Massons, J.; Parra, O.; Besses, C. Hematological disorders: A commonly unrecognized cause of acute stroke. Expert Rev. Hematol. 2016, 9, 891–901.
- Leng, T.; Xiong, Z.-G. Treatment for ischemic stroke: From thrombolysis to thrombectomy and remaining challenges. Brain Circ. 2019, 5, 8–11.
- Christophe, B.R.; Mehta, S.H.; Garton, A.L.A.; Sisti, J.; Connolly, E.S. Current and future perspectives on the treatment of cerebral ischemia. Expert Opin. Pharmacother. 2017, 18, 573–580.
- Uzdensky, A.B.; Demyanenko, S.V. Epigenetic mechanisms of ischemic stroke. Biochem. Suppl. Ser. A Membr. Cell Biol. 2019, 13, 289–300.
- Dhir, N.; Medhi, B.; Prakash, A.; Goyal, M.K.; Modi, M.; Mohindra, S. Pre-clinical to clinical translational failures and current status of clinical trials in stroke therapy: A brief review. Curr. Neuropharmacol. 2020, 18, 596–612.
- Shein, N.; Shohami, E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol. Med. 2011, 17, 448–456.
- Baltan, S.; Morrison, R.S.; Murphy, S.P. Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapy 2013, 10, 798–807.
- Fessler, E.; Chibane, F.; Wang, Z.; Chuang, D.-M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain Damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120.
- Merson, T.D.; Bourne, J.A. Endogenous neurogenesis following ischaemic brain injury: Insights for therapeutic strategies. Int. J. Biochem. Cell Biol. 2014, 56, 4–19.
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71.
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189.
- Jhelum, P.; Karisetty, B.C.; Kumar, A.; Chakravarty, S. Implications of epigenetic mechanisms and their targets in cerebral ischemia models. Curr. Neuropharmacol. 2017, 15, 815–830.
- Demyanenko, S.; Uzdensky, A. Epigenetic alterations induced by photothrombotic stroke in the rat cerebral cortex: Deacetylation of histone h3, Upregulation of Histone Deacetylases and Histone acetyltransferases. Int. J. Mol. Sci. 2019, 20, 2882.
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031.
- Schweizer, S.; Meisel, A.; Märschenz, S. Epigenetic mechanisms in cerebral ischemia. Br. J. Pharmacol. 2013, 33, 1335–1346.
- Hu, Z.; Zhong, B.; Tan, J.; Chen, C.; Lei, Q.; Zeng, L. The emerging role of epigenetics in cerebral ischemia. Mol. Neurobiol. 2016, 54, 1887–1905.
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23.
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198.
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the crossroads between different types of HDAC inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 2019, 20, 2415.
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749.
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42.
- Baltan, S.; Bachleda, A.; Morrison, R.S.; Murphy, S.P. Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl. Stroke Res. 2011, 2, 411–423.
- Lin, T.-N.; Kao, M.-H. Histone deacetylases in stroke. Chin. J. Physiol. 2019, 62, 95–107.
- Demyanenko, S.V.; Dzreyan, V.A.; Uzdensky, A.B. The expression and localization of histone acetyltransferases HAT1 and PCAF in neurons and astrocytes of the photothrombotic stroke-induced penumbra in the rat brain cortex. Mol. Neurobiol. 2020, 57, 3219–3227.
- Xu, Y.; Wang, Q.; Chen, J.; Ma, Y.; Liu, X. Updating a strategy for histone deacetylases and its inhibitors in the potential treatment of cerebral ischemic stroke. Dis. Markers 2020, 2020, 1–8.
- Bardai, F.H.; Verma, P.; Smith, C.; Rawat, V.; Wang, L.; D’Mello, S.R. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J. Neurosci. 2013, 33, 11833–11838.
- Lin, Y.-H.; Dong, J.; Tang, Y.; Ni, H.-Y.; Zhang, Y.; Su, P.; Liang, H.-Y.; Yao, M.-C.; Yuan, H.-J.; Wang, D.-L.; et al. Opening a New Time Window for Treatment of Stroke by Targeting HDAC2. J. Neurosci. 2017, 37, 6712–6728.
- Montgomery, R.L.; Hsieh, J.; Barbosa, A.C.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. USA 2009, 106, 7876–7881.
- Kelly, R.D.; Cowley, S. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749.
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344.
- Noh, K.-M.; Hwang, J.-Y.; Follenzi, A.; Athanasiadou, R.; Miyawaki, T.; Greally, J.M.; Bennett, M.V.L.; Zukin, R.S. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc. Natl. Acad. Sci. USA 2012, 109, E962–E971.
- Hwang, J.-Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200.
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60.
- Tang, Y.; Lin, Y.; Ni, H.; Dong, J.; Yuan, H.; Zhang, Y.; Liang, H.; Yao, M.; Zhou, Q.; Wu, H.; et al. Inhibiting histone deacetylase 2 (HDAC2) promotes functional recovery from stroke. J. Am. Hear. Assoc. 2017, 6, 007236.
- Lin, Y.; Yao, M.; Wu, H.; Dong, J.; Ni, H.; Kou, X.; Chang, L.; Luo, C.; Zhu, D. HDAC2 (Histone deacetylase 2): A critical factor in environmental enrichment-mediated stroke recovery. J. Neurochem. 2020, 155, 679–696.
- Demyanenko, S.; Neginskaya, M.; Berezhnaya, E. Expression of class I histone deacetylases in ipsilateral and contralateral hemispheres after the focal photothrombotic infarction in the mouse brain. Transl. Stroke Res. 2018, 9, 471–483.
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447.
- Chen, Y.-T.; Zang, X.-F.; Pan, J.; Zhu, X.-L.; Chen, F.; Chen, Z.-B.; Xu, Y. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin. Exp. Pharmacol. Physiol. 2012, 39, 751–758.
- Xia, Y.; Wang, J.; Liu, T.-J.; Yung, W.A.; Hunter, T.; Lu, Z. C-jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol. Cell 2007, 25, 219–232.
- Soriano, F.; Hardingham, G.E. In cortical neurons HDAC3 activity suppresses RD4-dependent SMRT export. PLoS ONE 2011, 6, e21056.
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 2012, 287, 35444–35453.
- Panteleeva, I.; Rouaux, C.; Larmet, Y.; Boutillier, S.; Loeffler, J.-P.; Boutillier, A.-L. HDAC-3 Participates in the repression of e2f -dependent gene transcription in primary differentiated neurons. Ann. NY Acad. Sci. 2004, 1030, 656–660.
- Takase, K.; Oda, S.; Kuroda, M.; Funato, H. Monoaminergic and neuropeptidergic neurons have distinct expression profiles of histone deacetylases. PLoS ONE 2013, 8, e58473.
- Bolger, T.A.; Zhao, X.; Cohen, T.J.; Tsai, C.-C.; Yao, T.-P. The neurodegenerative disease protein Ataxin-1 antagonizes the neuronal survival function of myocyte enhancer factor-2. J. Biol. Chem. 2007, 282, 29186–29192.
- Majdzadeh, N.; Wang, L.; Morrison, B.; Bassel-Duby, R.; Olson, E.N.; D’Mello, S.R. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev. Neurobiol. 2008, 68, 1076–1092.
- Chen, B.; Cepko, C.L. HDAC4 regulates neuronal survival in normal and diseased retinas. Science 2009, 323, 256–259.
- Price, V.; Wang, L.; D’Mello, S.R. Conditional deletion of histone deacetylase-4 in the central nervous system has no major effect on brain architecture or neuronal viability. J. Neurosci. Res. 2013, 91, 407–415.
- Demyanenko, S.; Dzreyan, V.; Uzdensky, A. Overexpression of HDAC6, but not HDAC3 and HDAC4 in the penumbra after photothrombotic stroke in the rat cerebral cortex and the neuroprotective effects of α-phenyl tropolone, HPOB, and sodium valproate. Brain Res. Bull. 2020, 162, 151–165.
- Bolger, T.A. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 2005, 25, 9544–9553.
- Yuan, H.; Denton, K.; Liu, L.; Li, X.-J.; Benashski, S.; McCullough, L.; Li, J. Nuclear translocation of histone deacetylase 4 induces neuronal death in stroke. Neurobiol. Dis. 2016, 91, 182–193.
- Koppel, I.; Timmusk, T. Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115.
- McCullough, L.D.; Tarabishy, S.; Liu, L.; Benashski, S.; Xu, Y.; Ribar, T.; Means, A.; Li, J. Inhibition of calcium/calmodulin-dependent protein kinase kinase β and calcium/calmodulin-dependent protein kinase IV is detrimental in cerebral ischemia. Stroke 2013, 44, 2559–2566.
- Kassis, H.; Shehadah, A.; Chopp, M.; Roberts, C.; Zhang, Z.G. Stroke induces nuclear shuttling of histone deacetylase 4. Stroke 2015, 46, 1909–1915.
- Demyanenko, S.; Berezhnaya, E.; Neginskaya, M.; Rodkin, S.; Dzreyan, V.; Pitinova, M. Class II histone deacetylases in the post-stroke recovery period—expression, cellular, and subcellular localization—Promising targets for neuroprotection. J. Cell. Biochem. 2019, 120, 19590–19609.
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC 4/5- HMGB 1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542.
- Lee, H.-A.; Song, M.-J.; Seok, Y.-M.; Kang, S.-H.; Kim, S.-Y.; Kim, I. histone deacetylase 3 and 4 complex stimulates the transcriptional activity of the mineralocorticoid receptor. PLoS ONE 2015, 10, e0136801.
- Cho, Y.; Sloutsky, R.; Naegle, K.; Cavalli, V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 2013, 155, 894–908.
- Schneider, J.W.; Gao, Z.; Li, S.; Farooqi, M.; Tang, T.-S.; Bezprozvanny, I.; Frantz, U.E.; Hsieh, J. Small-molecule activation of neuronal cell fate. Nat. Chem. Biol. 2008, 4, 408–410.
- Wei, J.-Y.; Lu, Q.-N.; Li, W.-M.; He, W. Intracellular translocation of histone deacetylase 5 regulates neuronal cell apoptosis. Brain Res. 2015, 1604, 15–24.
- Li, N.; Yuan, Q.; Cao, X.-L.; Zhang, Y.; Min, Z.-L.; Xu, S.-Q.; Yu, Z.-J.; Cheng, J.; Zhang, C.; Hu, X.-M. Opposite effects of HDAC5 and p300 on MRTF-A-related neuronal apoptosis during ischemia/reperfusion injury in rats. Cell Death Dis. 2017, 8, e2624.
- Gu, X.; Fu, C.; Lin, L.; Liu, S.; Su, X.; Li, A.; Wu, Q.; Jia, C.; Zhang, P.; Chen, L.; et al. miR-124 and miR-9 mediated downregulation of HDAC5 promotes neurite development through activating MEF2C-GPM6A pathway. J. Cell. Physiol. 2018, 233, 673–687.
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell–cell interactions. Trends Cell Biol. 2008, 18, 291–297.
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179.
- Khoury, N.; Koronowski, K.B.; Young, J.I.; Perez-Pinzon, M.A. The NAD+-dependent family of sirtuins in cerebral ischemia and preconditioning. Antioxid. Redox Signal 2018, 28, 691–710.
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635.
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116.
- She, D.T.; Jo, N.-G.; Arumugam, T. Emerging roles of sirtuins in ischemic stroke. Transl. Stroke Res. 2017, 8, 405–423.
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D’Mello, S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE 2008, 3, e4090.
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832.
- Gao, D.; Zhang, X.; Jiang, X.; Peng, Y.; Huang, W.; Cheng, G.; Song, L. Resveratrol reduces the elevated level of MMP-9 induced by cerebral ischemia–reperfusion in mice. Life Sci. 2006, 78, 2564–2570.
- Hisahara, S.; Chiba, S.; Matsumoto, H.; Tanno, M.; Yagi, H.; Shimohama, S.; Sato, M.; Horio, Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 15599–15604.
- Eng, F.; Ewijaya, L.; Etang, B.L. SIRT1 in the brain—connections with aging-associated disorders and lifespan. Front. Cell. Neurosci. 2015, 9, 64.
- Jiménez, M.H.; Hurtado, O.; Cuartero, M.; Ballesteros, I.; Moraga, A.; Pradillo, J.; McBurney, M.W.; Lizasoain, I.; Moro, M.A. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke 2013, 44, 2333–2337.
- Hattori, Y.; Okamoto, Y.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Kinoshita, M.; Ihara, M. SIRT1 attenuates severe ischemic damage by preserving cerebral blood flow. NeuroReport 2015, 26, 113–117.
- Li, Z.; Pang, L.; Fang, F.; Zhang, G.; Zhang, J.; Xie, M.; Wang, L. Resveratrol attenuates brain damage in a rat model of focal cerebral ischemia via up-regulation of hippocampal Bcl-2. Brain Res. 2012, 1450, 116–124.
- Luo, J.; Nikolaev, A.; Imai, S.-I.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 2001, 107, 137–148.
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015.
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.; A. Sinclair, D. Acetylation of the C terminus of Ku70 by CBP and PCAF controls bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638.
- Jin, Q.; Yan, T.; Ge, X.; Sun, C.; Shi, X.; Zhai, Q. Cytoplasm-localized SIRT1 enhances apoptosis. J. Cell. Physiol. 2007, 213, 88–97.
- Demyanenko, S.; Gantsgorn, E.; Rodkin, S.; Sharifulina, S. Localization and expression of Sirtuins 1, 2, 6 and plasticity-related proteins in the recovery period after a photothrombotic stroke in mice. J. Stroke Cerebrovasc. Dis. 2020, 29, 105152.
- Sugino, T.; Maruyama, M.; Tanno, M.; Kuno, A.; Houkin, K.; Horio, Y. Protein deacetylase SIRT1 in the cytoplasm promotes nerve growth factor-induced neurite outgrowth in PC12 cells. FEBS Lett. 2010, 584, 2821–2826.
- Krey, L.; Lühder, F.; Kusch, K.; Czech-Zechmeister, B.; Könnecke, B.; Outeiro, T.F.; Trendelenburg, G. Knockout of Silent Information Regulator 2 (SIRT2) preserves neurological function after experimental stroke in mice. Br. J. Pharmacol. 2015, 35, 2080–2088.
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 2011, 20, 3986–3996.
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444.
- Nahhas, F.; Dryden, S.C.; Abrams, J.; Tainsky, M.A. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 2007, 303, 221–230.
- Skoge, R.H.; Ziegler, M. SIRT2 inactivation reveals a subset of hyperacetylated perinuclear microtubules inaccessible to HDAC6. J. Cell Sci. 2016, 129.
- Xie, X.Q.; Zhang, P.; Tian, B.; Chen, X.Q. Downregulation of NAD-dependent deacetylase SIRT2 protects mouse brain against ischemic stroke. Mol. Neurobiol. 2016, 54, 7251–7261.
- Nie, H.; Hong, Y.; Lu, X.; Zhang, J.; Chen, H.; Li, Y.; Ma, Y.; Ying, W. SIRT2 mediates oxidative stress-induced apoptosis of differentiated PC12 cells. NeuroReport 2014, 25, 838–842.
- Favero, G.; Rezzani, R.; Rodella, L.F. Sirtuin 6 nuclear localization at cortical brain level of young diabetic mice: An immunohistochemical study. Acta Histochem. 2014, 116, 272–277.
- Tennen, R.I.; Chua, K.F. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 2011, 36, 39–46.
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.-C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free. Radic. Biol. Med. 2012, 52, 928–936.
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104.
- Cardinale, A.; De Stefano, M.C.; Mollinari, C.; Racaniello, M.; Garaci, E.; Merlo, D. Biochemical characterization of Sirtuin 6 in the brain and its involvement in oxidative stress response. Neurochem. Res. 2014, 40, 59–69.
- Shao, J.; Yang, X.; Liu, T.; Zhang, T.; Xie, Q.R.; Xia, W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 2016, 7, 281–290.
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.-S.; Chen, P.-S.; Chuang, D.-M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901.
- Ren, M.; Leng, Y.; Jeong, M.; Leeds, P.R.; Chuang, D. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: Potential roles of histone deacetylase inhibition and heat shock protein induction. J. Neurochem. 2004, 89, 1358–1367.
- Wang, Z.; Tsai, L.-K.; Munasinghe, J.; Leng, Y.; Fessler, E.B.; Chibane, F.; Leeds, P.; Chuang, D.-M. Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 2012, 43, 2430–2436.
- Yu, I.T.; Park, J.-Y.; Kim, S.H.; Lee, J.-S.; Kim, Y.-S.; Son, H. Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 2009, 56, 473–480.
- Hao, Y.; Creson, T.; Zhang, L.; Li, P.; Du, F.; Yuan, P.; Gould, T.D.; Manji, H.K.; Chen, G. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J. Neurosci. 2004, 24, 6590–6599.
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. NeuroMolecular Med. 2009, 11, 28–42.
- Park, M.J.; Sohrabji, F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J. Neuroinflammation 2016, 13, 1–14.
- Kim, H.J.; Leeds, P.; Chuang, D.-M. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 2009, 110, 1226–1240.
- Langley, B.; D’Annibale, M.A.; Suh, K.; Ayoub, I.; Tolhurst, A.; Bastan, B.; Yang, L.; Ko, B.; Fisher, M.; Cho, S.; et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21waf1/cip1 in cell cycle-independent neuroprotection. J. Neurosci. 2008, 28, 163–176.
- Qi, X.; Hosoi, T.; Okuma, Y.; Kaneko, M.; Nomura, Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol. Pharmacol. 2004, 66, 899–908.
- Jaworska, J.; Zalewska, T.; Sypecka, J.; Ziemka-Nalecz, M. Effect of the hdac inhibitor, sodium butyrate, on neurogenesis in a rat model of neonatal hypoxia–ischemia: Potential mechanism of action. Mol. Neurobiol. 2019, 56, 6341–6370.
- Patnala, R.; Arumugam, T.; Gupta, N.; Dheen, S.T. HDAC inhibitor sodium butyrate-mediated epigenetic regulation enhances neuroprotective function of microglia during ischemic stroke. Mol. Neurobiol. 2016, 54, 6391–6411.
- Tang, F.; Guo, S.; Liao, H.; Yu, P.; Wang, L.; Song, X.; Chen, J.; Yang, Q. Resveratrol enhances neurite outgrowth and synaptogenesis via sonic hedgehog signaling following oxygen-glucose deprivation/reoxygenation injury. Cell. Physiol. Biochem. 2017, 43, 852–869.
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol. 2006, 70, 1876–1884.
- Li, S.; Lu, X.; Shao, Q.; Chen, Z.; Huang, Q.; Jiao, Z.; Huang, X.; Yue, M.; Peng, J.; Zhou, X.; et al. Early histone deacetylase inhibition mitigates ischemia/reperfusion brain injury by reducing microglia activation and modulating their phenotype. Front. Neurol. 2019, 10, 893.
- Langley, B.; Brochier, C.; Rivieccio, M.A. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke 2009, 40, 2899–2905.
- Aune, S.E.; Herr, D.J.; Kutz, C.J.; Menick, D.R. Histone deacetylases exert class-specific roles in conditioning the brain and heart against acute ischemic injury. Front. Neurol. 2015, 6, 145.
- Pickell, Z.; Williams, A.M.; Alam, H.B.; Hsu, C.H. Histone deacetylase inhibitors: A novel strategy for neuroprotection and cardioprotection following ischemia/reperfusion injury. J. Am. Heart Assoc. 2020, 9, e016349.
- Shukla, S.; Tekwani, B.L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol. 2020, 11, 537.
More