Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Andrés Morales and Version 3 by Vivi Li.
Fritillaria bulbs are used in Traditional Chinese Medicine to treat several illnesses. Peimine (Pm), an anti-inflammatory compound from Fritillaria, is known to inhibit some voltage-dependent ion channels and muscarinic receptors, but its interaction with ligand-gated ion channels remains unexplored.
- peimine
- traditional Chinese medicine
- anti-inflammatory compound
1. Introduction
The scientific interest in Traditional Chinese medicines (TCMs) has bloomed in the last decades, as they provide a broad source of compounds of putative clinical relevance. The active ingredients of TCM plants include: (i) alkaloids, such as peimine (Pm), found in Fritillaria bulbs (Fb), which are commonly used to treat cough and asthma [1][2][1,2], (ii) terpenoids, as ginsenoides from Panax ginseng, which might reverse multidrug resistance of certain chemotherapeutic drugs [3], (iii) phenols and flavonoids (polyphenolic compounds), which are common in medicinal plants and own strong antioxidant and anti-inflammatory activities [4], tannic acid-related gallotannins and polyphenols, which are relatively abundant in green tea, inhibit TMEM16A, a calcium-activated chloride channel [5] and some flavonoids, as quercetin or genistein, act as positive allosteric modulators of α7 nicotinic receptors [6], and (iv) other compounds, as cinnamaldehyde, obtained from cinnamon cortex, which is known to activate TMEM16A [7].
Pm, also known as verticine, has been related to diverse therapeutic actions [8], including: (i) anti-inflammatory and analgesic, (ii) antitumor, inhibiting proliferation of human leukemia or lung cancer, (iii) expectorant, (iv) sedative, besides its analgesic action, (v) antihypertensive, (vi) a blocker of voltage-dependent ion channels, including both Na+ and K+ channels, and (vii) antimuscarinic, mainly acting on the M2 receptor subtype. Of note, Pm has a very low oral bioavailability, mainly because of its limited water solubility, and its intestinal absorption seems to take place through active transport, being pH-dependent [9]. In fact, by using Caco-2 cell monolayers, it has been estimated that the percentage of Pm absorbed would not exceed 2%, which means a poor absorption [9]. Consequently, it is expected that Pm actions should be elicited by high-affinity binding to specific targets, likely involved in inflammation, pain, smooth-muscle relaxation, sedation, or exocrine secretion. To date, the main effects of Pm have been attributed to different mechanisms of action, including: (i) use-dependent inhibition of voltage-dependent Nav1.7 channels, which would promote pain relief [10]; (ii) inhibition of voltage-dependent K+ channels, including the Kv1.3 expressed in lymphocytes and other non-excitable cells; in fact, Kv1.3. blockade by Pm has been related to its anti-inflammatory action [10]. Furthermore, Pm inhibits hERG (human ether-a-go-go related gene) channels by promoting their inactivation [11]. Remarkably, hERG channels play a critical role in the repolarization of myocardial cells and, thus, in the cardiac excitability; (iii) M2 muscarinic receptor inhibition, which might account for asthma amelioration, since M2 receptors are involved in airway smooth-muscle contraction [12]; (iv) angiotensin-converting enzyme inhibition, which would contribute to its antihypertensive action [13]; (v) enhancement of intracellular Ca2+ concentration, promoting phosphorilation of both Ca2+/calmodulin-dependent protein kinase II (CaMKII) and c-Jun N-terminal kinase (JNK), which inhibits growth and motility of cancer cells [14]; (vi) inhibition of P-glycoprotein expression in drug-resistance cells, reducing the ability of cancer cells to survive from chemotherapy [3]. At present, Fb are regarded as therapeutic herbs, despite that one of its main active compounds, Pm, blocks hERG channels (IC50 circa 40 µM) and, hence, it might trigger severe alterations in cardiac excitability. The fact that Fritillaria intake is yet considered safe, strongly suggests that its bioactive compounds, as Pm, must bind to specific targets with affinities much higher than those required to mediate the inactivation of hERG channels. However, presently, most putative therapeutic actions of Pm are elicited at concentrations similar to, or above, those required to block hERG channels, which suggests that Pm should act on additional molecular targets. Interestingly, as far as we know, it has not been yet assessed if Pm interacts with ligand-gated ion channels. This is rather remarkable since, for instance, nicotinic acetylcholine (ACh) receptors (nAChRs) are widely expressed in non-neuronal cells, including macrophages [15], and different subtypes of nAChRs have been involved in pain, inflammation, lung cancer, and proliferation of smooth-muscle and endothelial cells [15][16][15,16].
The nAChR belongs to the “Cys-loop” family of receptors, which are involved in fast synaptic transmission. Although all nAChRs are pentameric proteins, there is a large heterogeneity in their structures. Thus, whereas some nAChRs are homomeric, as those constituted by α7-10 subunits, others are heteromeric, containing specific combinations of α1-6, β1-4, γ, ε, and δ subunits. This large assortment of structural conformations of nAChRs accounts for a huge heterogeneity of functional and pharmacological properties of nAChRs. To date, the best characterized nAChR is the muscle type, located at postsynaptic membranes of both skeletal muscle fibers and electrocytes of some electric fishes, such as Torpedo. This nAChR is composed of 2α1, 1β1, 1δ, and either 1ε (adult-type) or 1γ (fetal-type) subunits, disposed delimiting a central channel pore [17][18][19][20][17,18,19,20].
2. IACh Blockade by Pm
The membrane conductance of oocytes either uninjected or bearing microtransplanted nAChRs was unaffected by bathing the cell with Pm (up to 100 µM) while holding the membrane potential at −60 mV. By contrast, co-application of ACh (10 µM) together with Pm (0.02–100 µM) to microinjected oocytes reversibly reduced the peak-amplitude (Ip) of IACh, in a dose-dependent manner (Figure 1B), following a sigmoid function (Figure 1C). At Pm concentrations over 0.1 µM, the extent of IACh inhibition measured 20 s after Ip (Iss) was greater than that corresponding to Ip values. In this way, the IC50 and nH values (see equation 1) for the Ip were 2.9 µM (confidence interval (CI), 2.0–4.3 µM; n = 5–16, N = 3–8) and 0.7 ± 0.1, respectively, whereas the dose-inhibition curve for the Iss displayed a lower IC50 (1.2 µM; CI 0.9–1.5 µM) but a similar slope (0.8 ± 0.1; the same cells and donor frogs as above; Figure 1C). Most likely, this lower IC50 for Iss is because of the enhancement of nAChR desensitization by Pm (see below).
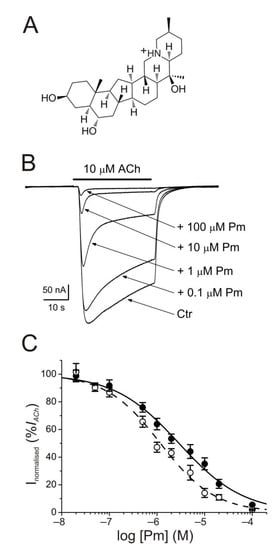
Figure 1. Inhibition of acetylcholine-elicited currents (IAChs) by peimine (Pm). (A) Molecular structure of Pm showing the charged nitrogen. (B) Superimposed IAChs evoked by 10 µM ACh either alone (Ctr) or co-applied with different Pm concentrations, as stated on the right. Note that Pm accelerates IACh decay when applied at concentrations of 0.1 μM or above. Hereafter, unless otherwise stated, the holding potential was −60 mV, downward deflections represent inward currents and the bars above recordings indicate the timing of drug application. (C) Pm concentration-IACh inhibition relationship. IACh amplitudes at their peak (Ip; filled symbols) and at their steady state (Iss, measured 20 s after the peak; open symbols) were normalized to the IACh evoked by ACh alone and plotted against the logarithm of Pm concentration. Solid and dashed lines are sigmoid curves fitted to Ip and Iss data, respectively. Error bars indicate SEM. Each point is the average of 5–16 oocytes from 3–8 frogs.
The specificity of Pm effects on muscle-type nAChR blockade was assessed by testing its effects on GABA subtype A receptors (GABAARs), which belong to the same Cys-loop family. For these experiments, oocytes were microinjected with rat brain synaptosomal membranes, which allowed the incorporation of GABAARs into the oocyte membrane [21]. These cells were later challenged with GABA (1 mM) either alone or together with Pm (up to 100 µM). Both IGABA amplitude and kinetics were rather unaffected by the presence of Pm (Supplementary Figure S1), in contrast to the marked inhibition of muscle-type nAChRs by Pm.
3. Competition Assays
The pharmacological profile of nAChR inhibition by Pm was determined by superfusing oocytes with ACh at different concentrations (3, 10, 30, 100 µM, and 1 mM) either alone or together with Pm at a concentration close to its IC50 (3 µM). Co-application of 10 µM ACh with 3 µM Pm halved Ip, as expected from the computed IC50 (Figure 1C). Similar percentages of Ip inhibition were found when Pm was co-applied with different ACh concentrations (Figure 2A–C), suggesting that Pm is acting as a non-competitive blocker. Notably, the percentages of Iss inhibition attained by co-applying Pm (3 µM) with different ACh concentrations were higher than those corresponding to the Ip inhibition (Figure 2A,C). Furthermore, the percentage of Iss inhibition increased significantly as the ACh concentration augmented (Figure 2C). This ACh concentration-dependence of Iss inhibition might be related to the enhancement of nAChR desensitization by Pm (see below), since the rate of desensitization is known to be dependent on ACh concentration [22][23][22,23].
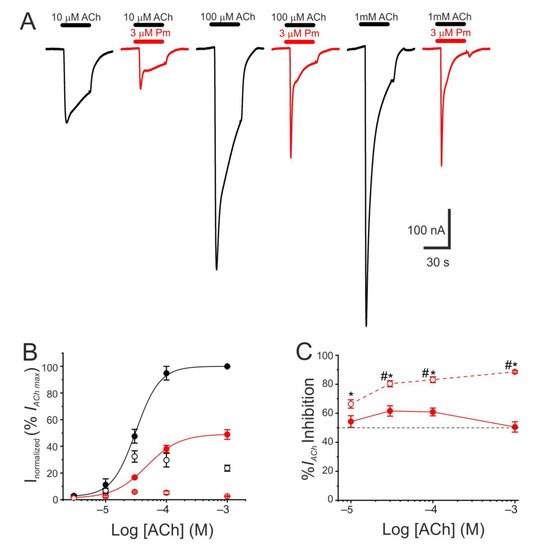
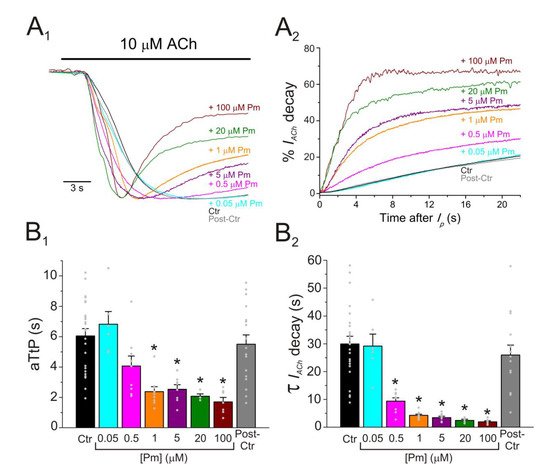
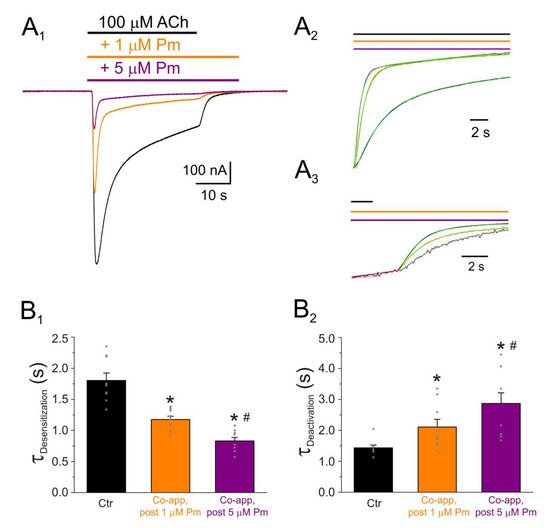 Figure 4. IACh decay and deactivation kinetics depend on Pm concentration. (A1,A2,A3) Representative IAChs elicited by 100 µM ACh either alone (black recording) or together with 1 (orange) or 5 (purple) µM Pm (A1). Pm superfusion remained 12 s after ACh washout (as indicated by the application bars). These recordings were normalized to either the same Ip, to better compare their IACh decay (A2), or the same Iss, to facilitate comparisons of deactivation kinetics (A3). (B1,B2) Column bar plots displaying the effect of 1 (orange) or 5 µM (purple) Pm on the IACh decay time-constant (τDesensitization; (B1)) and the deactivation kinetics (τDeactivation; (B2)), as compared to control IAChs (in the presence of ACh alone; black). (*) indicates significant differences with the control group (p < 0.05, paired t-test) and (#) indicates differences between 1 and 5 µM Pm groups (n = 9, N = 3; the same cells for all comparisons; p < 0.05, paired t-test). Notice that Pm accelerated the desensitization rate and slowed down the deactivation kinetics.
Figure 4. IACh decay and deactivation kinetics depend on Pm concentration. (A1,A2,A3) Representative IAChs elicited by 100 µM ACh either alone (black recording) or together with 1 (orange) or 5 (purple) µM Pm (A1). Pm superfusion remained 12 s after ACh washout (as indicated by the application bars). These recordings were normalized to either the same Ip, to better compare their IACh decay (A2), or the same Iss, to facilitate comparisons of deactivation kinetics (A3). (B1,B2) Column bar plots displaying the effect of 1 (orange) or 5 µM (purple) Pm on the IACh decay time-constant (τDesensitization; (B1)) and the deactivation kinetics (τDeactivation; (B2)), as compared to control IAChs (in the presence of ACh alone; black). (*) indicates significant differences with the control group (p < 0.05, paired t-test) and (#) indicates differences between 1 and 5 µM Pm groups (n = 9, N = 3; the same cells for all comparisons; p < 0.05, paired t-test). Notice that Pm accelerated the desensitization rate and slowed down the deactivation kinetics.
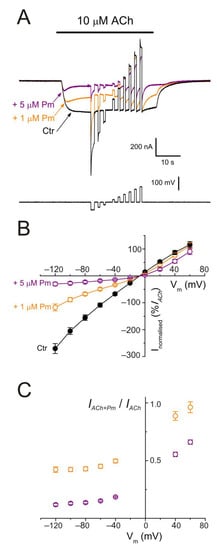
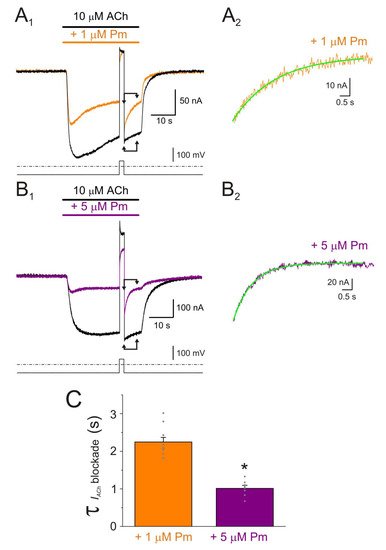 Figure 6. Kinetics of the voltage-dependent blockade of nAChR by Pm. (A1,B1) IAChs elicited by 10 µM ACh either alone (black recordings) or in the presence of 1 (orange; (A1)) or 5 µM (purple; (B1)) Pm, at −60 mV. A 2 s voltage jump to +40 mV was given at the IACh plateau to unplug the channel pore of the positively-charged Pm. Membrane leak-currents (recorded in the absence of ACh) have been subtracted. (A2,B2) Zooming in to the area indicated by the arrows in panels (A1,B1) (just after the voltage jump). The τ of the voltage-dependent blockade of nAChRs by Pm was determined by fitting an exponential function (green curve over the recording) to the net IACh decay. Before fitting, the smaller and slower IAChs evoked by ACh alone (black recordings of panels (A1,B1)) were subtracted from the IAChs in the presence of Pm. (C) Column-graph of τ values of the voltage-dependent blockade of nAChR by 1 and 5 µM Pm (same color code as in panels (A,B)). (*) indicates significant differences of τ values between both Pm concentrations (p < 0.05, t-test). Data are for 10 (N = 3) and 7 (N = 2) oocytes for 1 µM and 5 µM Pm, respectively.
Figure 6. Kinetics of the voltage-dependent blockade of nAChR by Pm. (A1,B1) IAChs elicited by 10 µM ACh either alone (black recordings) or in the presence of 1 (orange; (A1)) or 5 µM (purple; (B1)) Pm, at −60 mV. A 2 s voltage jump to +40 mV was given at the IACh plateau to unplug the channel pore of the positively-charged Pm. Membrane leak-currents (recorded in the absence of ACh) have been subtracted. (A2,B2) Zooming in to the area indicated by the arrows in panels (A1,B1) (just after the voltage jump). The τ of the voltage-dependent blockade of nAChRs by Pm was determined by fitting an exponential function (green curve over the recording) to the net IACh decay. Before fitting, the smaller and slower IAChs evoked by ACh alone (black recordings of panels (A1,B1)) were subtracted from the IAChs in the presence of Pm. (C) Column-graph of τ values of the voltage-dependent blockade of nAChR by 1 and 5 µM Pm (same color code as in panels (A,B)). (*) indicates significant differences of τ values between both Pm concentrations (p < 0.05, t-test). Data are for 10 (N = 3) and 7 (N = 2) oocytes for 1 µM and 5 µM Pm, respectively.
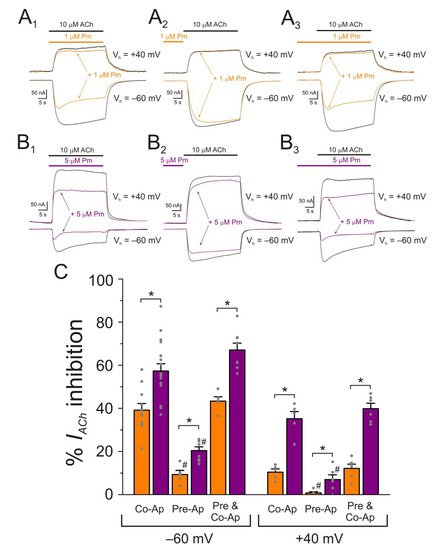 Figure 7. Effect of Pm-application timing and holding potential on nAChR blockade. (A1–A3) IAChs elicited at −60 mV (downward deflections) and at +40 mV (upward deflections) by co-application of 10 µM ACh and 1 µM Pm (A1), solely Pm pre-application before superfusing the agonist (A2) or Pm pre-application followed by its co-application with ACh (A3). (B1–B3) As in panels (A1–A3), but in the presence of 5 µM Pm instead of 1 µM. (C) Column graph shows the percentages of Ip inhibition by Pm when applied as indicated in panels (A1–A3,B1–B3), at −60 mV (on the left) and +40 mV (on the right). (*) indicates significant differences between Ip inhibition elicited by 1 and 5 µM Pm (p < 0.05, t-test). (#) denotes significant differences, for each Pm concentration, among the percentages of Ip inhibition elicited by ACh and Pm co-application and other Pm-application protocols, at the same holding potential (p < 0.05, ANOVA and Bonferroni t-test). Each column is the average of 5–11 and 5–17 oocytes, for −60 mV and +40 mV, respectively.
Figure 7. Effect of Pm-application timing and holding potential on nAChR blockade. (A1–A3) IAChs elicited at −60 mV (downward deflections) and at +40 mV (upward deflections) by co-application of 10 µM ACh and 1 µM Pm (A1), solely Pm pre-application before superfusing the agonist (A2) or Pm pre-application followed by its co-application with ACh (A3). (B1–B3) As in panels (A1–A3), but in the presence of 5 µM Pm instead of 1 µM. (C) Column graph shows the percentages of Ip inhibition by Pm when applied as indicated in panels (A1–A3,B1–B3), at −60 mV (on the left) and +40 mV (on the right). (*) indicates significant differences between Ip inhibition elicited by 1 and 5 µM Pm (p < 0.05, t-test). (#) denotes significant differences, for each Pm concentration, among the percentages of Ip inhibition elicited by ACh and Pm co-application and other Pm-application protocols, at the same holding potential (p < 0.05, ANOVA and Bonferroni t-test). Each column is the average of 5–11 and 5–17 oocytes, for −60 mV and +40 mV, respectively.
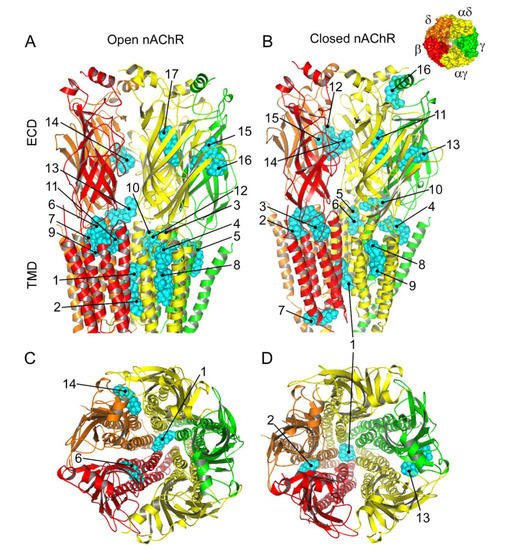 Figure 8. Predicted binding sites for Pm-nAChR complexes. (A,B) Lateral view of the nAChR displaying the main Pm (labelled in cyan) clusters bound to the open (A) and closed (B) conformations. The predicted loci are numbered consecutively, beginning in the transmembrane (TMD) and later in the extracellular (ECD) domain. (C, D) Top view of the nAChR (from the synaptic cleft) displaying representative Pm clusters binding to residues located within the channel pore, TMD, and ECD in the open (C) and closed (D) conformations. The inset, in the upper right corner, displays the nAChR subunits with their color code.
Our virtual docking and MD simulations used as a template the structure of Torpedo nAChRs in the open and closed conformations released by Unwin’s group [30][31][32,33]. However, a new structural model of the nAChR from Torpedo, at higher resolution and stabilized in the closed conformation by α-bungarotoxin, has been recently disclosed [32][34]. The new structural model (pdb entry 6UWZ) share large similarities with the Unwin’s model for the closed conformation, though there are certain differences between them. Mostly, they differ in the upper portion of the pore, which is more constricted in the new model, and in the δ subunit arrangement [33][35]. It seems that these discrepancies arise because of differences in the lipid matrix surrounding the nAChR. Actually, cholesterol interactions with the nAChR are apparently essential for stabilizing its structure and the absence of cholesterol (as in the model of Rahman et al. [32][34]) leads to a more compact arrangement of TM helices (displacement of helices circa 1–3 Å; [33][35]). Noticeably, the major lipid present in electroplax membranes rich in nAChRs is cholesterol [34][36] and purified nAChRs from T. marmorata and E. electricus interact preferentially with cholesterol rather than with either phospholipid monolayers or other sterols [35][37]. Moreover, nAChRs in native electroplax membranes are arranged as dimers, linked by their δ-subunits. This interaction between neighboring nAChRs might account for the differences in the δ-subunit between both structural models since dimers were reduced to monomeric receptors in the Rahman’s model. In fact, we chose for our structural studies Unwin’s models because of: (i) the structures for the open and closed conformations are available, (ii) the nAChR is present in their original membrane, and (iii) we have significant experience correlating structural and functional results using these commonly accepted models; actually, Unwin’s models have so far provided a coherent correlation with our functional results [21][28][27][36][21,25,26,28].
The virtual docking assays predicted Pm binding to the nAChR at different sites of the TMD and ECD in the open conformation. Most Pm clusters were located at the TMD, at inter- and intra-subunit crevices, although some of them located into the channel pore (Figure 89A,C). The binding energies estimated for these clusters were rather high (from −9.3 to −12.87 kcal/mol; see Supplementary Table S1), pointing out that Pm has a high affinity for the nAChR. Remarkably, MD simulations of nAChRs in the open conformation indicate that Pm binding to the nAChR either at the TMD (i.e., cluster 6) or at the ECD (i.e., cluster 14) markedly decreased both the volume and the number of water molecules at the hydrophobic gate region of the channel pore (Figure 911). Furthermore, virtual docking and MD assays pointed out that Pm also interacts with the nAChR in the resting conformation, binding to residues located at both the TMD and the ECD (Figure 89B,D and Figure 911E,F). Interestingly, the structural changes of the nAChR induced by Pm, as predicted by docking and MD assays, are in good agreement with the functional changes elicited by Pm on this receptor. Actually, the structural and functional results can be correlated as follows: (i) the high binding energies computed accounted for the high potency of Pm blocking nAChRs, (ii) Pm interaction with residues located within the channel pore should trigger the open-channel blockade, (iii) Pm binding to different sites at the nAChR might explain both its heterogeneity of actions on nAChRs and the effect-dependence on Pm concentration, and (iv) Pm binding to the nAChR in the closed conformation might underlie the blockade of resting nAChR. Consequently, the good correlation between structural simulations and electrophysiological results strongly suggests that Pm actually blocks nAChRs by the different aforementioned mechanisms.
Figure 8. Predicted binding sites for Pm-nAChR complexes. (A,B) Lateral view of the nAChR displaying the main Pm (labelled in cyan) clusters bound to the open (A) and closed (B) conformations. The predicted loci are numbered consecutively, beginning in the transmembrane (TMD) and later in the extracellular (ECD) domain. (C, D) Top view of the nAChR (from the synaptic cleft) displaying representative Pm clusters binding to residues located within the channel pore, TMD, and ECD in the open (C) and closed (D) conformations. The inset, in the upper right corner, displays the nAChR subunits with their color code.
Our virtual docking and MD simulations used as a template the structure of Torpedo nAChRs in the open and closed conformations released by Unwin’s group [30][31][32,33]. However, a new structural model of the nAChR from Torpedo, at higher resolution and stabilized in the closed conformation by α-bungarotoxin, has been recently disclosed [32][34]. The new structural model (pdb entry 6UWZ) share large similarities with the Unwin’s model for the closed conformation, though there are certain differences between them. Mostly, they differ in the upper portion of the pore, which is more constricted in the new model, and in the δ subunit arrangement [33][35]. It seems that these discrepancies arise because of differences in the lipid matrix surrounding the nAChR. Actually, cholesterol interactions with the nAChR are apparently essential for stabilizing its structure and the absence of cholesterol (as in the model of Rahman et al. [32][34]) leads to a more compact arrangement of TM helices (displacement of helices circa 1–3 Å; [33][35]). Noticeably, the major lipid present in electroplax membranes rich in nAChRs is cholesterol [34][36] and purified nAChRs from T. marmorata and E. electricus interact preferentially with cholesterol rather than with either phospholipid monolayers or other sterols [35][37]. Moreover, nAChRs in native electroplax membranes are arranged as dimers, linked by their δ-subunits. This interaction between neighboring nAChRs might account for the differences in the δ-subunit between both structural models since dimers were reduced to monomeric receptors in the Rahman’s model. In fact, we chose for our structural studies Unwin’s models because of: (i) the structures for the open and closed conformations are available, (ii) the nAChR is present in their original membrane, and (iii) we have significant experience correlating structural and functional results using these commonly accepted models; actually, Unwin’s models have so far provided a coherent correlation with our functional results [21][28][27][36][21,25,26,28].
The virtual docking assays predicted Pm binding to the nAChR at different sites of the TMD and ECD in the open conformation. Most Pm clusters were located at the TMD, at inter- and intra-subunit crevices, although some of them located into the channel pore (Figure 89A,C). The binding energies estimated for these clusters were rather high (from −9.3 to −12.87 kcal/mol; see Supplementary Table S1), pointing out that Pm has a high affinity for the nAChR. Remarkably, MD simulations of nAChRs in the open conformation indicate that Pm binding to the nAChR either at the TMD (i.e., cluster 6) or at the ECD (i.e., cluster 14) markedly decreased both the volume and the number of water molecules at the hydrophobic gate region of the channel pore (Figure 911). Furthermore, virtual docking and MD assays pointed out that Pm also interacts with the nAChR in the resting conformation, binding to residues located at both the TMD and the ECD (Figure 89B,D and Figure 911E,F). Interestingly, the structural changes of the nAChR induced by Pm, as predicted by docking and MD assays, are in good agreement with the functional changes elicited by Pm on this receptor. Actually, the structural and functional results can be correlated as follows: (i) the high binding energies computed accounted for the high potency of Pm blocking nAChRs, (ii) Pm interaction with residues located within the channel pore should trigger the open-channel blockade, (iii) Pm binding to different sites at the nAChR might explain both its heterogeneity of actions on nAChRs and the effect-dependence on Pm concentration, and (iv) Pm binding to the nAChR in the closed conformation might underlie the blockade of resting nAChR. Consequently, the good correlation between structural simulations and electrophysiological results strongly suggests that Pm actually blocks nAChRs by the different aforementioned mechanisms.
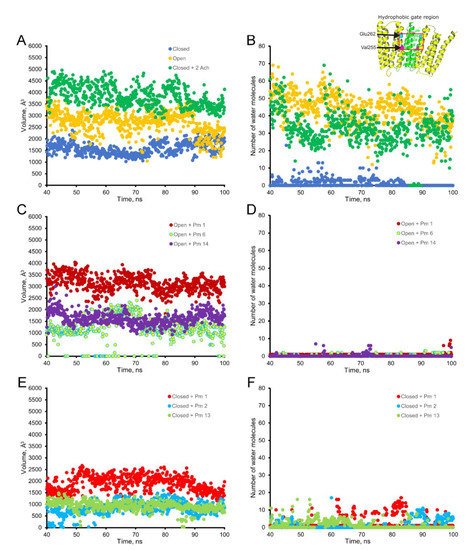
Figure 2. Pharmacological profile of nAChR blockade by Pm. (A) Representative IAChs evoked by ACh at 10 µM (left), 100 µM (middle), or 1 mM (right), either alone (black recordings) or together with 3 µM Pm (red recordings). (B) ACh concentration-IACh amplitude relationship when the cell was bathed with either just ACh (black color; filled symbols denote Ip values whereas open circles correspond to Iss) or ACh plus 3 µM Pm (red symbols). The EC50 values of the sigmoid curves fitting the experimental data were 31 µM (range 11–52 µM) and 47 µM (range 46–49 µM) for control and 3 µM Pm, respectively. (C) Percentage of Ip (solid symbols) and Iss (open symbols) inhibition elicited by 3 µM Pm when co-applied with the indicated ACh concentrations. (*) indicates significant differences between Ip and Iss inhibition, for each ACh concentration (p < 0.05, paired t-test). (#) indicates significant differences among Iss inhibition at 10 µM ACh and other concentrations (p < 0.05, ANOVA followed by Bonferroni t-test). Each point of panels B and C is the average of 4–14 cells from 1–2 donors.
4. Discussion
A broad number of medicinal plants have been used for centuries as therapeutic tools in TCM. However, neither the active compounds of many of these plants nor their mechanisms of action are yet well-understood. We have now studied the effect of Pm, an isosteroidal alkaloid considered one of the main bioactive molecules of Fb, on nAChRs. Remarkably, Pm decreased IACh in a dose-dependent manner, with an IC50 in the low micromolar range (circa 3 and 1 µM for Ip and Iss, respectively; Figure 1). These IC50s are markedly lower than the values previously reported for Pm blockade of voltage-dependent potassium channels. Thus, Pm IC50 for blocking Kv1.2 was 472 µM, it was 354 µM for Kv1.3 (142 µM if measured 150 ms after the current peak), and even much higher for Kv1.4 to Kv1.8 channels [10]; additionally, Pm inhibited the potassium channel hERG, with an IC50 of 44 µM, most likely by enhancing its inactivation [11]. Pm also blocked the Nav1.7 channel (IC50, 47 µM), demonstrating use-dependent inhibition, like the blocking mechanism of lidocaine on this channel [10]. Pm effects on hERG channels are particularly remarkable, since these channels play a key role in myocardial repolarization and, therefore, their inhibition might cause serious cardiac arrhythmias. Despite this, humans have used Fb as a therapeutic herb for centuries, being considered safe for consumption. Consequently, intake of Fb should not markedly affect the activity of hERG channels, despite that Pm, one of its main bioactive compounds, blocks these channels with an IC50 of 44 µM [11]. Nevertheless, Pm has a very low oral bioavailability [9] and thus its plasma concentration after Fb intake should be fairly low. Actually, Pm content in bulbs of Fritillaria ussuriensis and thunbergii ranged from 0.58 to 1.2 mg/g and oral administration of powder from these Fritillaria plants to dogs (1 g/kg) raised Pm plasma concentration to a maximum of 100–200 nM [24][30]. Accordingly, a similar Pm bioavailability was reported after oral administration of Fritillaria thunbergii extracts in rats, with peak plasma concentrations of Pm of roughly 100 nM [25][31]. These Pm concentrations are several orders of magnitude lower than the IC50s reported for sodium or potassium channels (including hERG), muscarinic receptors, or acethylcholinesterase [8]. Interestingly, submicromolar Pm concentrations elicit a significant inhibition of nAChRs (roughly 20%; Figure 1) and therefore this family of LGIC might be relevant targets of its actions. Of note, we have assessed Pm actions on muscle-type nAChRs, because they are the prototype member of this family of receptors, but Pm might have different affinities for related receptors, as the homomeric α7. In fact, α7 nAChRs are largely expressed in non-neuronal tissues, including macrophages, and exert powerful anti-inflammatory actions [16]. Moreover, other nAChR subtypes, such as α4β2 and/or α9α10, may also play a role in modulating inflammatory processes and even in chronic pain [15]. Noticeably, Pm displayed a differential affinity for different LGICs, even of the same family. Thus, whereas Pm inhibits muscle-type nAChRs with and IC50 close to 1 µM, GABAA receptors were almost not affected by Pm at concentrations up to 100 µM. Pm exerted a non-competitive inhibition on muscle-type nAChRs, since IAChs halved when co-applying Pm, at its IC50, with different ACh concentrations (Figure 2A,B). Several blockade mechanisms seem involved in this non-competitive inhibition of nAChRs by Pm, which is coherent with the multiple binding sites predicted by the docking simulations. First, open-channel blockade, as IACh inhibition by Pm was voltage-dependent, the more hyperpolarized the cell, the more pronounced the blockade (Figure 5). Actually, this is what would be expected for a positively charged molecule plugging the channel pore. In agreement with this, the docking assays predicted some Pm clusters located within the channel pore, both in the open and closed conformations (Figure 89). Thus, the open-channel blockade of nAChRs mediated by Pm resembles that mediated by some LAs, such as lidocaine [26][24] or tetracaine [27][26]. Nevertheless, the kinetics of open-channel blockade of nAChR elicited by Pm was rather slow. Thus, at −60 mV, the τ of open channel blockade elicited by 1 µM Pm (close to its IC50) was over 2 s and even by 5 µM Pm (eliciting roughly 70% of Iss blockade) the time constant was above 1 s (Figure 67). In contrast, at the same membrane potential, 0.7 µM tetracaine (close to its IC50) blocked open nAChRs with a time course of roughly 300 ms [27][26]. These differences in time constants between Pm and tetracaine are most likely related to their distinct molecular sizes (Pm molecular weight is over 60% greater than that of tetracaine). Second, Pm enhanced nAChR desensitization, as evidenced by: (i) acceleration of IACh decay when co-applying ACh with Pm at concentrations of 0.5 µM, or above (Figure 3A2,B2), and (ii) shortening of the IACh aTtp, which correlated with the acceleration of IACh decay (Figure 3A1,B1). Likewise, lidocaine decreased the IACh aTtp only at concentrations that enhanced IACh decay [26][24]. Furthermore, DMA, a lidocaine analog, both sped up IACh decay and shortened the aTtP [28][25]. By contrast, diethylamine (DEA), a lidocaine analog that mainly blocks nAChR by open channel blockade neither accelerates IACh decay nor decreases aTtP [21], and (iii) deceleration of IACh deactivation, which was dependent on Pm concentration and displayed a good correlation with the rate of IACh decay (Figure 4A, B). The deceleration of IACh deactivation when Pm remained in the solution strongly supports that Pm enhanced nAChR desensitization, since desensitized nAChRs display higher affinity for the agonist [22][27][29][22,26,27]. Third, Pm elicited the blockade of resting (closed) nAChRs. This effect was unraveled by applying Pm before challenging the cells with ACh alone. This protocol, which allowed Pm to act only on resting (closed) nAChRs, elicited a mild nAChR blockade, mostly at negative potentials. Actually, at positive potentials, IACh only decreased by Pm pre-application when rising its concentration to 5 µM (Figure 78). In agreement with this, IACh inhibition by 5 µM Pm was slightly higher when Pm was pre-applied and then co-applied with ACh than when solely co-applied with ACh (Figure 78C).Figure 3. Pm accelerates IACh decay and shortens the time to reach Ip. (A1,A2) Superimposed IAChs elicited by 10 µM ACh either alone (black and grey recordings) or together with different Pm concentrations (shown at right). IAChs were scaled to the same Ip amplitude to better compare the differences in time to reach Ip (aTtP; A1) and kinetics of IACh decay after Ip (A2). (B1,B2) Column graphs displaying Pm effects on aTtP (B1) and τ-values of IACh-decay (B2). (*) indicates significant differences among IAChs in presence of Pm (colored columns; same color code as in (A1,A2)) and their control values (Ctr, black column; p < 0.05, ANOVA and Bonferroni t-test). Note that post-control values (after Pm applications; grey column) were similar to control ones. Each point is the average of 4–24 cells (N = 3–12).
Figure 5. nAChR blockade by Pm is voltage-dependent. (A) IAChs evoked by 10 µM ACh alone (black recording) or in the presence of either 1 (orange) or 5 µM (purple) Pm when applying voltage pulses from −120 to +60 mV, as illustrated underneath. (B) Net i/v relationship of IAChs elicited by the protocol shown in (A). Black symbols are for control IAChs, whereas those evoked in the presence of Pm are drawn in either orange (+1 µM Pm) or purple (+5 µM Pm). Net IAChs were normalized as the percentage of their control IACh at −60 mV (n = 5–11; N = 2–3). (C) Plot displaying the IACh remnant after co-application of either 1 (orange) or 5 µM (purple) Pm (IACh+Pm), normalized to their control (IACh), versus the membrane potential (same cells as in (B)). Notice that 1 µM Pm, in contrast to 5 µM, did not significantly decrease IACh at +60 mV.
Figure 9. Empty volume (left panels) and number of water molecules (right panels) within the hydrophobic gate of the nAChR pore region (between Val255 and Glu262 of the alpha subunit; see inset in the upper right corner), through the period of 40 to 100 ns of MD simulations. Top panels show the empty volume (A) and the number of water molecules (B) at the hydrophobic gate region in control nAChR (in the absence of Pm), both in the open (yellow) and the closed (blue) conformations; also, it displays the effect of ACh on the closed conformation (green). Middle panels (C,D) display the effect of Pm on these parameters when located at representative sites of the nAChR in the open conformation: within the channel pore (Pm 1), at the TMD (Pm 6), and the ECD (Pm 14). Lower panels (E,F) demonstrate the effect of Pm at representative loci of the nAChR in the closed conformation: inside the channel (Pm 1), at TMD (Pm 2), and at ECD (Pm 13).
Since Fb has been used as therapeutic herb for thousands of years, there is a strong support for its beneficial effects and its weak (or lack) of toxicity. However, neither the identity of all its bioactive compounds nor their mechanisms of action are yet well known. Now, we report here that Pm, considered one of the main bioactive compounds from Fritillaria, exerts a powerful inhibition of muscle-type nAChRs, which, as far as we know, is the first report demonstrating that Pm might modulate LGICs, besides acting on other targets as voltage-dependent channels or metabotropic receptors. It remains to be unraveled if Pm might modulate other nAChRs, including the homomeric α7, which is broadly expressed in immune cells and has been related to powerful anti-inflammatory actions [16]. Furthermore, both mecamylamine, a non-competitive antagonist of α7 nAChRs, and 1-ethyl-4-(3-(bromo)phenyl)piperazine, which promotes α7 desensitization, reduce pro-inflammatory responses [37][38]. We have now demonstrated that Pm inhibits muscle-type nAChRs and it can be hypothesized that it might modulate, though in a different way or with a different potency, either α7 or other nAChRs. In this regard, lidocaine exerted similar inhibitory actions on muscle-type nAChRs [26][24] and on neuronal nAChRs expressed in autonomic-ganglia neurons [38][39]. Alternatively, it could be that different bioactive compounds from Fb accounted for its anti-inflammatory actions, as anthocyanin pigments, which are flavonoids present in Fb [39][40]. Noticeably, some flavonoids act as positive allosteric modulators of α7 nAChRs, although without affecting desensitization [6], and their enhancement of α7 activity has been proposed as a therapeutic strategy for inflammatory disorders [40][41].