Pluripotent stem cells (PSCs) hold great potential both in studies on developmental biology and clinical practice. Mitochondrial metabolism that encompasses pathways that generate ATP and produce ROS significantly differs between PSCs and somatic cells. Correspondingly, for quite a long time it was believed that the redox homeostasis in PSCs is also highly specific due to the hypoxic niche of their origin – within the pre-implantation blastocyst. However, recent research showed that redox parameters of cultivated PSCs have much in common with that of their differentiated progeny cells. Moreover, it has been proven that similar to somatic cells, maintaining the physiological ROS level is critical for the regulation of PSC identity, proliferation, differentiation, and de-differentiation.
1. Introduction
Oxygen is a critical factor for the existence of all aerobic organisms. Redox reactions, inherent in aerobic metabolism, are among the most abundant chemical interactions in living cells. Low molecular weight oxidants called reactive oxygen species (ROS) play a key role in these reactions. ROS are ions and neutral molecules, often in the form of free radicals, that are more reactive than molecular oxygen. In total, more than a dozen different substances belong to the ROS family—superoxide anion radical, hydrogen peroxide (H
2O
2), hydroxyl radical, singlet oxygen, etc. (a detailed list can be found in the review
[1]). At the onset of redox biology, ROS were considered to be only the toxic by-product of cellular respiration, but later it became clear that ROS play an important role in metabolic and signaling processes that regulate cell proliferation, differentiation, motility, and apoptosis
[1,2,3,4][1][2][3][4]. The sources of ROS are usually subdivided into mitochondrial and non-mitochondrial. The main producers of mitochondrial ROS are several enzymatic complexes of the mitochondrial electron transport chain (ETC)
[5[5][6],
6], whereas the main, but not single, non-mitochondrial ROS generator is a family of NADPH oxidases (NOXs)
[7]—in total, more than 40 enzymes are known to produce ROS
[1]. The main mediator of redox signaling is H
2O
2, which is enzymatically produced in different cellular compartments—either directly in reactions involving molecular oxygen or by dismutation of superoxide anion radical, also formed from O
2 [8]. In parallel with the intracellular generation of ROS, their rapid elimination constantly occurs, and enzymatic systems responsible for that compose the antioxidant defense system of the cell. ROS are removed by highly productive enzymes such as superoxide dismutases, peroxiredoxins, glutathione peroxidase, and catalase
[9]. These enzymes provide precise control of intracellular ROS, keeping them at a very low level—in particular, about 10
−9 M in the case of H
2O
2 and 10
−11 M in the case of superoxide anion radical
[1].
The situation when intracellular ROS level exceeds the physiological norm is called oxidative stress, or more specifically, ‘oxidative distress’
[10]. Under conditions of distress, high oxidizing ability of ROS causes extensive damage to cell macromolecules—proteins, lipids, and nucleic acids
[11]. In this regard, in the early studies of cellular redox homeostasis, the main attention was paid to the negative impact of oxidative distress induced by external factors or redox metabolism disorders. To cope with the oxidative stress conditions, it was proposed to use pharmacologic antioxidants, defined as substances that “delay, prevent or remove oxidative damage to a target molecule”
[12]. Later it was found that elevated ROS production accompanies and worsens the course of many human diseases, such as cardiovascular, neurodegenerative, oncological, and others. From that moment on, antioxidants were actively tested for use in therapy; however, surprisingly, clinical trials did not meet the expectations, often having no effect or leading to a worse prognosis
[13,14][13][14]. Failures in using antioxidants for clinical practice, together with the accumulated fundamental knowledge on cellular redox systems, have brought to the forefront studies of molecular mechanisms that ensure the maintenance of normal redox homeostasis as well as research on the regulatory role of ROS in metabolic and signaling processes. In contrast to the term ‘oxidative distress’, for the description of the physiological ROS level modulation that performs signaling or regulatory functions the term ‘oxidative eustress’ was introduced
[10,15][10][15].
The shift of the paradigm that took place in redox biology at the turn of the century proceeded in parallel with the development of stem cell research. The derivation of pluripotent embryonic stem cells (ESCs) from mouse (mESCs)
[16,17][16][17] and then from human (hESCs)
[18] at the very end of the 20th century became a turning point in stem cell and developmental biology, as well as in regenerative medicine. ESCs give rise to the formation of all tissues and organs of an organism. The proliferative and differentiating potential of these cells ensures the development of the embryo; therefore, the ability to study the properties of these cells and induce their differentiation into various cell types of three germ layers in vitro is of particular importance for both fundamental and practical aspects of biomedicine. The use of differentiated descendants of ESCs for the treatment of various human pathologies initially held a great promise
[19], but the question of histocompatibility remained open. Thus, further development of a technology for obtaining induced pluripotent stem cells (iPSCs) from adult cells by ectopic expression of only four transcription factors
[20,21][20][21] has once again spurred interest in the study of pluripotency.
Pluripotent stem cells (PSCs), both ESCs and iPSCs, have a number of unique characteristics that distinguish them from somatic cells: a clonal growth capacity, shortened proliferation cycle, high ability for DNA damage repair, and inability to activate the programs of cellular senescence, due to, among the other things, telomerase activity inherent in these cells
[18,22][18][22]. PSCs also have specific metabolic features corresponding to their initial localization within the blastocyst in the hypoxic conditions of the uterine cavity. While the mitochondrial electron transport chain (ETC) is the main energy source in somatic cells, PSCs rely to a greater extent on anaerobic glycolysis even when cultured at atmospheric oxygen levels
[23,24,25][23][24][25]. Their mitochondria possess morphological and functional differences in comparison to the mitochondrial network of differentiated cells
[24]. Since mitochondrial enzymes of ETC are one of the main sources of intracellular ROS, highlighting the peculiarities of PSC energy metabolism led to the concept that the redox homeostasis in PSCs significantly differs from that of their differentiated progeny cells and that PSCs can limit intracellular ROS production to minimize ROS-induced oxidative damage. However, recent studies have shown that PSCs have not only phenotypic but also metabolic plasticity and, therefore, are able to adjust their redox metabolism to the conditions of their microenvironment
[26,27][26][27]. These cells a priori have the capacity to exist not only in hypoxic conditions but also in normoxia (21% O
2)
[26] and even hyperoxia (>21% O
2)
[28]. Recently published data show that under conditions in vitro, redox parameters of PSCs have much in common with the redox characteristics of differentiated cells
[29,30][29][30]. Moreover, in PSCs, similarly to somatic cells, maintaining the physiological ROS level turned out to be critical for the regulation of their identity, proliferation, differentiation, and de-differentiation
[31].
2. Redox Homeostasis in Pluripotent Stem Cells
2.1. Quantification of the ROS Level in PSCs
The main characteristic of cell redox homeostasis is an intracellular ROS level, which is usually determined by assessing the ability of a whole cell (or its individual compartments) to oxidize redox probes. Fluorescent compounds (dyes or genetically encoded biosensors), prone to change their fluorescence upon oxidation, are generally used as such probes. Some of them (e.g., many biosensors
[32]) are specifically oxidized by individual ROS compounds, but for the most part, redox probes are capable of being oxidized by the variety of intracellular oxidants and are commonly used to assess the overall ROS level in cells
[33,34][33][34].
The first studies aimed to assess the level of ROS in pluripotent cells were carried out using the most common among all redox-sensitive dyes, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). H2DCFDA is originally fluorescently inactive but starts to emit intense fluorescence upon oxidation. This probe can react with various intracellular oxidants; however, it mostly is oxidized by H
2O
2 [34]. In 2004, it was found that the level of ROS in cultured mESCs appeared to be several times lower than that in their spontaneously differentiated progeny cells, as well as in embryonic mouse fibroblasts and mouse fibroblast line 3T3 cells
[28]. Since then, numerous studies have shown that both murine and human PSCs (ESCs, iPSCs) have very weak ability to oxidize H2DCFDA in comparison to their differentiated counterparts
[35,36,37][35][36][37]. Experiments using other ROS-sensitive dyes (peroxide-sensitive DHR 123
[35[35][38],
38], superoxide-sensitive DHE
[37], mitochondrial superoxide-sensitive MitoSOX
[35,38][35][38]) showed similar results. At first, these observations were explained by the peculiarities of the redox metabolism in PSCs. It is known that these cells originate from the preimplantation blastocyst, which is surrounded by an intrauterine fluid with an oxygen content of about 4%
[39,40][39][40]. Considering the hypoxic conditions in which PSCs exist in the body, a hypothesis about the low ability of pluripotent cells to generate ROS was formulated (discussed in
[41]). According to this hypothesis, low generation of ROS in PSCs allows preventing oxidative damage to cell proteins, lipids, and DNA. However, later on, more detailed studies of intracellular redox environment in hESCs carried out in comparison with their fibroblast-like descendants, as well as differentiated human cells of different nature and origin (lymphocytes, fibroblasts, mesenchymal stem cells, HeLa cells), have shown that the differences in the cell capacities to oxidize redox-sensitive probes depend strongly on the cell sizes
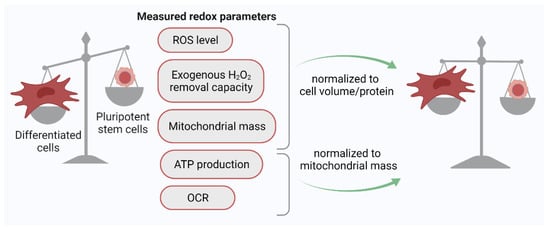
[30]. For the adequate comparison of the redox status of different cells using the H2DCFDA probe, it was proposed to use normalized parameters: the ratio of the H2DCFDA signal to the cell volume (biophysical normalization) or cell protein (biochemical normalization). The use of this approach showed that the ROS level evaluated on a per volume or per protein basis does not differ significantly between PSCs and differentiated human cells (
Figure 1). Thus, it eventually appeared that PSCs and their differentiated progeny cells are equally committed to generating ROS, at least under normoxic (21% O
2) culture conditions.
Figure 1. Normalization of key redox parameter values to cell protein/volume or mitochondrial mass leads to the leveling of these parameters between PSCs and their differentiated progeny cells. ROS, reactive oxygen species; OCR, oxygen consumption rate.
2.2. ROS Production in PSCs
2.2.1. Mitochondrial Activity in PSCs
Mitochondrial ROS generation is closely related to cell bioenergetics. Cellular energy is produced in the processes of multistage oxidation (dehydrogenation) of various substrates (sugars, proteins, lipids, amino acids) formed during the catabolism of nutrients consumed by the body. Some stages of this oxidation process occur in the cell cytoplasm, and others—inside mitochondria, but all of them are coupled to the production of ATP. Mitochondrial ATP synthesis is accompanied by generation of ROS. The ATP and ROS outputs in cellular mitochondria depend thus on the activity of mitochondria
[42].
The first stage of glucose breakdown (glycolysis), which takes place in the cytoplasm, does not require oxygen. During glycolysis, ATP is synthesized by substrate phosphorylation (i.e., by attaching to ADP a phosphate group after it is cleaved from phosphate-containing compounds). In the course of glycolysis, pyruvate is formed, which can be further converted into lactate under anaerobic conditions. Alternatively, pyruvate can be transported into mitochondria where, after decarboxylation, it associates with coenzyme A. The product of this reaction, Acetyl-CoA, participates in the next aerobic, intra-mitochondrial stage of glucose decomposition in the tricarboxylic acid (TCA) cycle, during which the acetyl residues (CH
3CO-) are oxidized to carbon dioxide (CO
2). Along with glucose degradation products, the TCA cycle can be fueled by breakdown products of other substrates (lipids, amino acids)
[42]. Compounds generated in the TCA cycle, in turn, support the work of the electron transport chain, a series of protein complexes found on the inner mitochondrial membrane. ETC produces ATP through oxidative phosphorylation (OXPHOS) reactions, in which ATP synthase catalyzes the addition of inorganic phosphate to ADP. It is worth noting that besides its role in oxidative catabolism of carbohydrates and fatty acids, the TCA cycle also provides precursors for many biosynthetic pathways, including precursors for amino acid and nucleotide synthesis
[25]. The production of mitochondrial ROS (mitoROS) is interlinked with bioenergetic metabolism via the OXPHOS. Electron leak from ETC leads to the generation of a highly reactive metabolite of molecular oxygen, superoxide anion (O
2−·), formed by one-electron reduction of O
2. O
2−·, in turn, is rapidly dismutated to H
2O
2 by two dismutases—Cu/Zn-superoxide dismutase (SOD1) in mitochondrial intermembrane space and Mn-superoxide dismutase (SOD2) in mitochondrial matrix. In the reactions of H
2O
2 with transition metal ions, the hydroxyl radical (OH·) is formed. In spite of the fact that free radical ROS are highly reactive, their lifetime is extremely short, while H
2O
2 is a much more stable and long-lived molecule.
First studies devoted to the bioenergetics of PSCs showed that these cells in comparison with their differentiated progeny cells produce a small amount of ATP, have a low rate of oxygen consumption, and also have a low mitochondrial mass and immature mitochondrial morphology
[24,29,43][24][29][43]. In addition, glycolysis, producing lactate, was shown to be highly active in PSCs even in the presence of oxygen. These data were at first interpreted as evidence of the insignificant contribution of OXPHOS processes to the bioenergetics of PSCs. This interpretation correlated well with the hypothesis about low oxidative activity in PSCs due to the hypoxic conditions in their original niche that dominated in the first decade of the 2000s. However, in further studies, this concept has undergone significant adjustments. In 2011, it was shown that the ratio of the mitochondrial mass to the total protein and the ratio of the oxygen consumption rate to the mitochondrial mass are almost equivalent between normal human dermal fibroblasts and human PSCs (both ESCs and iPSCs)
[29]. The data presented showed that, despite the morphological features of the mitochondrial network of PSCs and the active use of the glycolytic pathway for ATP synthesis, these cells possess functioning complexes of the mitochondrial respiratory chain and actively consume O
2 [29]. Later, these observations were supported by metabolic flux analysis, performed by Turner et al.
[26]. The results confirmed the high glycolytic nature of hESCs but, in addition, revealed a high activity of the TCA cycle in cellular mitochondria. The analysis showed that substrates used in this cycle by PSCs are mainly obtained not from glycolysis but using an alternative way—the process of catabolism of amino acids, in particular glutamine. In addition, the authors compared the metabolic characteristics of hESCs cultured at 2 and 21% O
2 and found high metabolic plasticity of PSCs. They demonstrated that under different oxygen conditions, the activity of the OXPHOS pathways in PSCs can change, and at low oxygen content, hESCs can increase the activity of glycolysis to meet the total energy requirement of the cell. The results of further studies
[44] confirmed that human PSCs (ESC and iPSC) rely both on glycolysis and glutamine oxidation for ATP generation, and hPSC viability critically depends on the presence of glutamine. Moreover, later it was found that metabolic shifts from glucose to glutamine oxidation and vice versa mediate the regulation of pluripotent cell identity. In-depth studies of both mouse and human PSCs have shown that pluripotency is not a discrete state but a wide spectrum of states, characterized by different potency and distinct metabolic profiles
[45]. Terms ‘naïve’ and ‘primed’ pluripotency have been introduced, which allows to specify cells that are capable or incapable, respectively, of incorporation into a developing blastocyst generating chimeric embryos. Moreover, within the ‘naïve’ PSC population, a sub-population of cells with enhanced pluripotency (so-called ground state of ‘naïve’ pluripotency) was identified. Recently, Vardhana and colleagues
[46] showed that transition towards this state increased the fraction of TCA cycle intermediates generated from glucose-derived carbons while decreasing the fraction of TCA cycle intermediates derived from glutamine.
Thus, the conducted studies have proven that for the production of ATP, PSCs rely on both oxygen-dependent and -independent metabolism to meet high requirements for energy production and substrates for anabolism needed for their high proliferative and functional activity. The idea of exclusively anaerobic metabolism with low mitochondrial activity has not been confirmed.
2.2.2. Non-Mitochondrial ROS Production in PSCs
In cells of various types, non-mitochondrial ROS are produced by plenty of redox-active enzymes (xanthine oxidase, nitric oxide synthase, etc.). Between them, NADPH oxidases (NOXs) are considered to be the main and best-studied sources of non-mitochondrial ROS. NOXs is a group of transmembrane complexes consisting of NOX1-5 and DUOX1-2 that are able to transport electrons from NADPH to oxygen, generating either superoxide anion radical, which can be further transformed into H
2O
2 due to the activity of SOD enzymes, or directly H
2O
2. NOX1-3 complexes produce O
2−· and interact with two membrane subunits (gp91-phox or its homologs, and p22-phox) that form the catalytic core of NOX, several cytosolic subunits (p47-phox, p67-phox, p40-phox), and the G-protein Rac, which are required for their assembly and activation
[47]. Unlike NOX1-3, NOX4 generates H
2O
2 and interacts only with p22phox; therefore, it is considered a constitutively active isoform that is regulated at the level of transcript expression (reviewed in
[48]). Since the superoxide anion dismutation rate is much higher than the reaction rate between superoxide and thiol groups of intracellular proteins, H
2O
2 is mainly considered to perform the signaling functions in the cell
[49], being able to oxidize the cysteine residues of signal transduction proteins. Interestingly, simple NOX homologs were first discovered in early eukaryotes, in slime mold, and fungi
[50]. A large-scale search for catalytic NOX subunits in unicellular and multicellular organisms showed the presence of these enzymes only in the latter, indicating the importance of NOX in ensuring the functioning of complex organisms and the coordination of intercellular signaling
[51,52][51][52]. In mammals, NADPH oxidases can be found within the plasma membrane (NOX1-5 and DUOX1-2), mitochondrial membrane (NOX4), the endoplasmic reticulum (NOX2, NOX4, and NOX5), and nuclear membrane (NOX4 and NOX5). Due to the specific subcellular localization of different NADPH oxidases, ROS production is compartmentalized, leading to modulation of intracellular redox signal cascades
[53]. The main function of NOXs is considered to be the regulation of multiple redox-dependent processes, such as proliferation, cell death, calcium signaling, cell differentiation, and reprogramming. NOX-produced ROS can increase intracellular calcium concentration by activating calcium channels, as well as directly oxidize some transcription factors such as NF-kB, HIF-1α, FOXOs, Nrf2, and p53
[54]. Most of these functions have been found for the first time in somatic cells; however, recently
[55] it has been shown that NOX2 and NOX4 contribute to pluripotency maintenance and self-renewal of mouse iPSs (miPSCs) (see
Section 2). Using the qPCR method, Kang and colleagues showed that miPSCs highly expressed
NOX2 and
NOX4, while the mRNA levels of other NOX family members were much lower (
NOX1, DUOX1, DUOX2) or undetectable (
NOX3 and
NOX5). In addition, a number of other works
[56,57,58][56][57][58] have shown that the expression of NADPH oxidases is temporarily increased during the differentiation of PSCs.
2.3. ROS Elimination in PSCs
All cells have a powerful system of antioxidant defense that allows precise control of intracellular ROS levels and a quick response to their threatening increases. Antioxidant defense system is an extensive group of both enzymatic and non-enzymatic substances. First-line defense enzymes are members of a superoxide dismutase (SOD) family. In addition to the already mentioned manganese SOD2 (localized in the mitochondrial matrix) and copper-zinc SOD1 (working in the cytoplasm, nucleus, and mitochondrial intermembrane space), cells also produce copper-zinc SOD3. The latter is secreted into the extracellular space and is anchored to the extracellular matrix and cell surface. SOD family provides dismutation of the superoxide produced by the mitochondrial and non-mitochondrial sources to H
2O
2. In turn, H
2O
2 scavenging is performed by several enzymatic systems. The first enzyme is catalase, although having a highly efficient reduction capacity for H
2O
2, it is localized mainly in peroxisomes and used for controlling the balance of H
2O
2 in these organelles
[59]. One more potent H
2O
2 scavenger is glutathione peroxidase (GPX), a member of the cytosolic oxidoreductase family that inactivates H
2O
2 with the use of two molecules of glutathione (GSH), whose thiol groups donate their electrons to H
2O
2, forming a disulfide bond (GSSG). The reduction of oxidized glutathione occurs due to the activity of the glutathione reductase enzyme (GSR), which uses NADPH as a substrate
[60]. Another highly efficient H
2O
2 detoxification enzyme is peroxiredoxin (PRX). Recent evidence suggests that PRX is not only capable of neutralizing H
2O
2, but it also enables relaying H
2O
2-derived oxidizing equivalents to other proteins, participating thus in the redox signaling events
[61]. PRX, in turn, can be reduced by thioredoxins (TRX), a family of small proteins that are also actively involved in maintaining the redox homeostasis of a cell. In similarity to GSH, the presence of cysteine residues in the TRX structure allows the reduction of not only PRX but also many other intracellular proteins, as well as the regulation of signaling cascades by direct interaction with redox-active members of various signaling pathways
[62].
PSCs, being parental cells assuring formation of all tissues of an organism, are equipped with an effective antioxidant system; however, the expression pattern of antioxidant genes differs from that revealed in the differentiated progenies of PSCs. Early studies of both murine and human ESCs and iPSCs have shown that expression of some antioxidant genes (
GPX2-4,
SOD2,
GSR), as well as accumulation of some antioxidant proteins (SOD2, catalase), is markedly reduced during PSC differentiation
[28,35,36,38][28][35][36][38]. Basing on these observations, it was suggested that pluripotent cells possess a highly potent antioxidant system, more effective than that of their differentiated descendants. However, several studies, aimed to compare the transcriptome profile of PSCs and their differentiated progenies or various cells from adult tissues, did not reveal antioxidant enzyme genes in the lists of differentially expressed genes
[63,64][63][64]. Similarly, genetic screenings or transcriptome profiling did not find principal antioxidant enzymes in the sets of genes that were demonstrated to be crucial for initiation and maintenance of pluripotency
[65,66,67][65][66][67]. In addition, functional tests for the PSC resistance to oxidative stress did not confirm the hypothesis about the enhanced antioxidant protection of these cells. The comparison of the rate constants of the H
2O
2 elimination between hESCs, their differentiated offspring, and adult mesenchymal stem cells showed that hESCs eliminate exogenous H
2O
2 even slower than any of the two differentiated cells listed above. However, when normalizing this constant not per cell, but per cell protein, the values became equal for hESCs and their differentiated progenies, indicating the same functional activity of their antioxidant systems
[30]. Like many other redox parameters of PSCs (such as low ROS level, oxygen consumption, mitochondrial mass), the poor ability of these cells to eliminate exogenous H
2O
2 appears to be related to their small size, which results in fewer H
2O
2 scavengers per cell in comparison to PSC differentiated counterparts.
2.4. Oxidative Stress Response in PSCs
The levels of ROS that exceed the physiological norm (‘oxidative distress’) are known to cause a strong damaging effect on cells. The fate of cells that survived oxidative stress depends on many factors: the potency of antioxidative defense, the ability of a cell to repair the damage, signaling pathways that shape stress response programs, etc. For example, in response to genotoxic impacts (including oxidative stress), transformed cells usually initiate apoptosis programs, while many normal cells of an adult organism, such as fibroblasts and mesenchymal stem cells, are prone to activating premature senescence
[68,69][68][69]. Predisposition to the certain stress response program determines the viability of cells after damaging factor withdrawal.
When studying the activity of DNA damage repair response in murine PSCs, Saretzki et al. found that radiation-induced DNA strand break repair is superior in mESCs in comparison to differentiated mESC progenies or mouse embryonic fibroblasts
[28]. mESCs repaired DNA faster than 3T3 mouse fibroblasts and, when cultured under hyperoxia (40% O
2), only slightly reduce their proliferation rate, while mouse embryonic fibroblasts stopped dividing. It was found also that many genes involved in stress response (heat shock genes,
BMI1,
ERCC4) decreased their expression during mESC differentiation
[28]. Another study
[70] also showed that, in comparison to somatic cells, both human ESCs and iPSCs possess high levels of DNA repair proteins (RAD51, Ku70, XLF, DNA LigIIIα, XRCC1, and PARP1) that participate in the double-strand break, single-strand break, and base excision repair pathways. Using functional tests (plasmid-based repair assay), Fan et al. showed that both hESCs and human iPSCs (hiPSCs) demonstrated elevated efficacy of nonhomologous end-joining, one of the main pathways for DNA double-strand breaks repair
[70].
However, when assessing the cytotoxic effect of exogenous H
2O
2 on hESCs and their differentiated offspring, the cytotoxic dose (estimated as the number of H
2O
2 moles per one cell) turned out to be ten times lower in hESCs, and when normalized to a cellular protein, which allows taking into account the difference in the cell volume—two times lower
[30]. Thus, despite the efficient system of DNA damage repair in PSCs, their resistance to the cytotoxic effect of oxidative stress turned out to be low. One of the reasons for this phenomenon was found in studies that showed both mESCs and hESCs quickly and efficiently eliminate oxidatively damaged cells by apoptosis, while their differentiated counterparts do not die, but undergo premature senescence
[71,72][71][72].
Thus, even though many parameters of redox homeostasis (overall ROS level and mitochondrial mass normalized per cell protein, oxygen consumption rate normalized per mitochondrial mass, activity of redox metabolism pathways, potency of the antioxidant system) turned out to be similar in PSCs and their differentiated progenies, the response of pluripotent cells to oxidative stress is fundamentally different from that of differentiated cells. PSCs turned out to be highly sensitive to the oxidative load due to their small size, but simultaneously they are more protected from the damaging effects of stress due to their ability to effectively repair DNA breaks and eliminate damage at the level of cell population.
3. Redox Signaling in Pluripotent Stem Cells
A lot of proteins participating in intracellular signaling cascades have thiol-rich cysteine residues in their structure, including in active centers. During oxidation, thiol groups lose hydrogen atoms and form disulfide bonds (or “disulfide bridges”) between their sulfur atoms that lead to a change in the conformation of the signaling protein and ensures signal transduction
[49]. It is important to note that the formation of disulfide bonds is a reversible process. The reduction of thiol groups occurs due to the activity of thiol–disulfide exchange enzymes, which mainly belong to glutathione- and thioredoxin-dependent enzymatic systems. Thus, the mechanism of redox signaling is a kind of switching between active/inactive protein states where ROS play the role of “fingers” pressing the switches. Different subcellular localization of sources and short lifetimes of ROS lead to compartmentalization and spatiotemporal distribution of intracellular redox signals, which results in the timely activation of strictly defined signaling pathways. PSCs perform two main functions: (1) they actively proliferate to maintain their population, and (2) they are able to differentiate into various cell types of the three germ layers on cue. A number of studies confirm the involvement of ROS in the regulation of the self-renewal process and differentiation of PSCs, as well as induction of pluripotency.