Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Manoj Govindarajulu and Version 2 by Rita Xu.
The widespread increase in multiple severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) variants is causing a significant health concern in the United States and worldwide. These variants exhibit increased transmissibility, cause more severe disease, exhibit evasive immune properties, impair neutralization by antibodies from vaccinated individuals or convalescence sera, and reinfection.
- SARS-CoV-2
- COVID-19
- variants of concern
- vaccines
1. Introduction
The commonly known severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the coronavirus disease 2019 (COVID-19) that initially appeared by public reports in December 2019 in China and then spread worldwide, causing a pandemic outbreak throughout 2020 and 2021. As of 24th September 2021, COVID-19 has led to approximately 230 million confirmed cases and caused over 4.7 million deaths worldwide. In the United States, there have been around 42 million confirmed cases of COVID-19, with 677,323 deaths [1].
Viral variants result from mutations during viral replication. A mutation is described as any change, such as a substitution, deletion, or addition, in a genetic sequence of a virus compared to the normal sequence. Coronaviruses are positive, single-stranded RNA viruses resembling a crown appearance under a microscope [2]. The mutation rate is slow compared to other common viruses, such as influenza [3]. This means SARS-CoV-2 is less likely to experience mutational changes, such as antigenic drift and antigenic shift responsible for altering the virus composition that leads to differences in infectivity, transmission, and disease severity. As COVID-19 spreads across the world, the virus naturally mutates to form new variants that can either be more or less infectious than the previous form depending on the altered composition. Some of the mutations, especially those occurring at the Spike (S) protein, could affect the entry of the virus into the target cells and the efficacy of the antibody protection. Specifically, mutations occurring in the Receptor-Binding Domain (RBD) of the S protein are of high significance as most vaccines and neutralizing antibodies target the RBD [4]. Other mutations in the S protein, such as one occurring at the N-terminal Domain (NTD), could impair the capability of the neutralizing antibodies as well [5]. With more studies, the impact of mutations occurring in other regions of the genome will be determined.
The D614G mutation in the S protein documented in the early part of the pandemic is found in almost every sequence worldwide. This mutation is characterized by the replacement of aspartic acid with glycine at position 614 of the S protein and influences viral infectivity [6]. Higher levels of viral RNA were noted in the patients, indicating high viral load and potential for higher infectivity [7]. As the transmission of the virus continued, several new variants with multiple mutations have emerged globally [8]. The Center for Disease Control (CDC), in collaboration with SARS-CoV-2 Interagency Group (SIG), classify SARS-CoV-2 variants into variants of concern (VOC), variant of interest (VOI), and variants of high consequence depending on the threat level they pose to the public’s health, as described in the following sections [9].
2. Nomenclature of SARS-CoV-2 Variants
The naming of the SARS-CoV-2 variants is based on Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) or interchangeably referred to as Pango lineage nomenclature [10]. According to the nomenclature, there are two major lineages, namely, A and B, at the root of the phylogeny of SARS-CoV-2. Lineage A viruses, for instance, the Wuhan/WHO4/2020 sequence sampled in January 2020, share two nucleotides (positions 8782 in ORF1ab and 28144 in ORF8) with the closest known bat viruses (RaTG13 and RmYN02). In comparison, lineage B, such as the Wuhan-Hu-1 strain sampled in December 2019, display different nucleotides at the above-mentioned sites. The additional SAR-CoV-2 genomes, which descend from either lineage A or lineage B, are designated a numerical value, for example, lineage A.1 or lineage B.2. Furthermore, these lineages (A.1 or lineage B.2) can act as predecessors for virus lineages that emerge in other geographical areas or at different time points, and these are designated with two sublevels, for instance, A.1.1. These designations can proceed for a maximum of three sublevels (e.g., A.1.1.1), after which new descendent lineages are given a letter (in English alphabetical sequence from C, so A.1.1.1.1 would become C.1 and A.1.1.1.2 would become C.2). These descendent lineages should show phylogenetic evidence of emergence from an ancestral lineage into another geographically distinct population, implying substantial forward transmission in that population. The following criteria are used to determine phylogenetic evidence for a new lineage:
-
Exhibits one or more shared nucleotide differences from the ancestral lineage
-
Comprises at least five genomes with >95% of the genome sequenced
Table 1. Classification of SARS-CoV-2 Variants.
| Pango Lineage and Corresponding WHO Naming | Attributes * | ||||
|---|---|---|---|---|---|
|
| ||||
| Variants of high consequences |
|
|
- Exhibits at least one shared nucleotide change among genomes within the lineage
- Demonstrate a bootstrap value >70% for the lineage-defining node.
As of September 2021, lineage B and its sub-lineage B.1 appears to be the most prevalent worldwide. In the United States, there are four circulating variants, with B.1.1.7 being the most common. The other variants include B.1.351, P.1 (B.1.1.28.1), and B.1.617.2 (the B.1.427 and B.1.429 variants have been de-escalated due to low prevalence). These variants appear to spread more easily and quickly than other variants, leading to more cases of COVID-19. The CDC categorizes SARS-CoV-2 variants into different groups regarding the potential for causing severe disease leading to morbidity and/or mortality, significant infectivity rate, or decreased response to SARS-CoV-2 antibodies generated from a previous infection or vaccination [11]. The different variants have been categorized into variants of interest (VOI), variants of concern (VOC), or variants of high consequence (VOHC) in the United States (Table 1
| Variants of Interest |
|
| ||||
| Variants of concern | ||||||
|
|
The classification is as per the Centers for Disease Control and Prevention (CDC) dated 17th September 2021. * As per the CDC definition. ^ The World Health Organization (WHO) named it the Lambda variant on 14th June 2021. # The World Health Organization (WHO) included the Mu variant as a variant of interest on 30th August 2021.
The World Health Organization (WHO) revised the naming system for SARS-CoV-2 (both VOC and VOI) based on the Greek alphabet, such as “Alpha”, “Beta” or “Gamma”. This system has the advantage of referring to the variants more quickly in a simplified scientific language, especially for non-scientists, national authorities, media, and others. Additionally, it avoids identifying them by the countries where they were identified first; thereby, preventing stigmatization of a country for detecting and reporting variants. The variants using the new WHO nomenclature are found in Table 1.
3. SARS-CoV-2 Genome
The genome of SARS-CoV-2 consists of a single-stranded positive-sense RNA consisting of 5′-UTR (untranslated region) and a poly(A)-tail at 3′-UTR, both of which assumes a structure similar to the mRNA of host cells (Figure 1). The proteins of SARS-CoV-2 consist of two large polyproteins: ORF1a and ORF1ab (that proteolytically cleave to form 16 nonstructural proteins), major transmembrane Spike (S) glycoprotein, membrane (M), nucleocapsid (N) protein, and small envelope (E) protein, and at least six accessory proteins: ORF3a, ORF6, ORF7a, ORF7b, ORF8a, and ORF8b, which follows a typical 5′-3′ order of appearance [12].
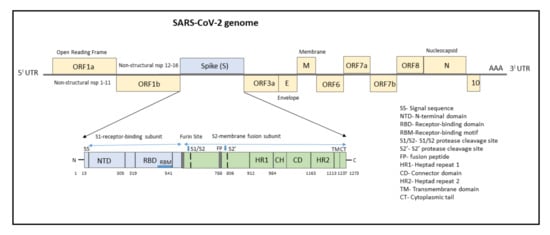
Figure 1. Structure of SARS-CoV-2 genome with domain structure of the Spike protein.
The ORF1a/b encodes a polyprotein termed polyprotein1a (pp1a), which corresponds to nonstructural proteins NSP1 to NSP11, and pp1b consisting of NSP12 to NSP16. Several functional domains of NSPs have been studied, notably the 3C-like cysteine proteinase (3CLpro, nsp5), RNA-dependent RNA polymerase (RdRp, nsp12), nidovirus RdRp-associated nucleotidyltransferase (N terminal of nsp12), helicase (Hel, nsp13), and exonuclease (ExoN, nsp14) [12][13][12,13]. Other nsps have a role in suppressing host cells, immunological suppression, and other functions. ORF1a Nsps are critical for controlling genome expression, while ORF1b Nsps are important for replication.
The structural RNA genome transcribes into the S protein with a size of 180–200 kDa, which plays a role in virus entry into host cells by binding to the human angiotensin-converting enzyme 2 (ACE2) receptor. The S protein consists of 1273 amino acids and consists of a signal sequence (aa 1–13), S1 subunit (14–685 residues), and S2 subunit (686–1273 residues), as shown in Figure 1.
The signal sequence consisting of 13 aa has high hydrophobic residues and helps in guiding the S protein to its membrane destination. The S1 subunit consists of the N-terminal domain (14–305) and receptor-binding domain (319–541). The N-terminal domain (NTD) plays a role in attachment, and mutations at NTD confer reduced sensitivity to neutralizing antibodies, which makes SARS-CoV-2 more permissive for deleterious escape mutations in the RBD. Neutralizing antibodies from both infected patients and vaccinated individuals target RBD and NTD. For instance, NTD-targeting antibodies bind to NTD to form an NTD/antibody complex, thereby preventing conformational changes in the S protein and block membrane fusion and viral entry [14]. The RBD plays a critical role in binding to the host cell ACE2 receptor in the region of aminopeptidase N. Studies indicate that the SARS-CoV-2 RBM has more residues that directly interact with the ACE2 receptor than the SARS-RBD [15]. Hence, mutations in the key residues in this region play an important role in enhancing the interaction with ACE2. The RBD functions to interact with human angiotensin-converting enzyme II (ACE2) and facilitates virus entry into host cells. The RBD-targeting neutralizing antibodies bind directly to S Protein RBD and compete for the ACE2 receptor resulting in neutralization of the virus [14].
The S2 subunit, which contributes to membrane fusion, consists of the fusion peptide (788–806), heptapeptide repeat sequence 1 (912–984), HR2 (1163–1213), TM domain (1213–1237), and cytoplasm domain (1237–1273). The furin-like cleavage domain is the site at which S protein gets cleaved into two parts, the S1 subunit and S2 subunit. SARS-CoV-2 possesses multiple furin cleavage sites, thereby increasing the probability of being cleaved by the host furin-like proteases. This induced cell–cell fusion to form syncytium to facilitate viral spread from one cell to another, thereby increases the chances of infectivity [16]. An additional cleavage site, referred to as the S2′site, is cleaved by the host TMPRSS2, where S2 is cleaved into FP and S2′domains. Hence, furin cleavage plays an important role in viral assembly, whereas TMPRSS2 cleavage triggers membrane fusion, syncytium formation, and viral entry into a target cell [17].
After the S protein gets cleaved, the fusion peptide (FP) undergoes a conformational change and inserts into the host membrane and anchors inside. The TM and CD stabilize the trimeric structure during the process of viral fusion and forms an anchor inside the virion [18]. Additionally, FP has been shown to mediate membrane fusion by disrupting and linking lipid bilayers of the host cell membrane [19]. Once the distance between the viral and host membrane is shortened, the HR1 and HR2 undergo conformational changes and mediates viral fusion and entry of the S2 subunit by bringing the viral envelope and host cell membrane close to one another [20].
Since the S protein is the main antigenic component of SARS-CoV-2, neutralizing antibodies targeting the S protein can induce protective immunity against the infection. Antibodies targeting various regions of the S protein have different mechanisms in preventing SARS-CoV-2 infection. For instance, monoclonal antibodies (mAbs) and nanobodies (Nbs) targeting the RBD form RBD/mAb or RBD/Nb complexes, which inhibit the binding of the RBD to ACE2, thereby prevent the entry of SARS-CoV-2 into the host cells. Conversely, mAbs targeting the NTD form the NTD/mAb complex and prevent conformational changes in the S protein and block membrane fusion and viral entry. However, RBD-targeting antibodies are more potent than the antibodies targeting other regions (such as NTD) of the S protein, but they might exhibit reduced efficacy in inhibiting multiple virus strains [14].
The envelope (E) protein, being the smallest structural protein, with a molecular weight of 8–12 kDa, possesses three important domains viz. (N)-terminus, transmembrane domain (TMD), and (C)-terminus. The (C)-terminal domain (motif DLLV) binds to human PALS1, a tight junction-associated protein, which is essential for the establishment and maintenance of epithelial polarity in mammals [21]. The E protein is involved in pathogenesis, virus assembly, and release [22]. Additionally, in cooperation with the M protein, it mediates host immune responses by the activation of NLRP3 inflammasome and PDZ binding function via its C-terminal domain.
The membrane (M) protein is a 25–30 kDa O-linked glycoprotein, binds to structural proteins, such as nucleocapsid, and promotes viral assembly, and increases its virulence [23]. The nucleocapsid (N) protein is responsible for RNA packaging, virus particle releasing, and the ribonucleoprotein core forming process. Mutations in the N protein pose a potential diagnostic risk as most commercially available antigenic rapid diagnostic tests detect the presence of the N protein [24].
The ORF3a located between the S and E genes is important for regulating apoptosis and inflammatory responses in the infected cells [25][26][25,26]. Additionally, ORF3a activates the innate immune signaling receptor NLRP3 inflammasome and causes tissue inflammation and cytokine production [27]. The ORF6 is important for increasing the viral infectivity by suppressing interferon production and interferon signaling [28]. The ORF7a and ORF7b are transmembrane proteins important for structural integrity, while the ORF8 of SARS-CoV-2 mediates the immune evasion by interacting with major histocompatibility complex molecules class I (MHC-I) and down-regulating the surface expression of MHC-I in various cells [29]. ORF9b associates with an adaptor protein, TOM70, and thereby suppresses IFN-I-mediated antiviral responses [30]. The SARS-CoV-2 ORF10 protein interacts with multiple human proteins expressed in the lung tissues and is found to maintain disease transmissibility [31]. Additionally, apart from viral replication, the above-mentioned accessory proteins play a role in host immune escape [32][33][32,33].